El raquitismo hipofosfatémico ligado al cromosoma X (XLHR, por sus siglas en inglés) es el más común de varios trastornos hereditarios y se caracteriza por la pérdida renal de fosfato, que da lugar a huesos débiles o blandos. Anteriormente conocida como "raquitismo resistente a la vitamina D", el XLHR no se considera actualmente una verdadera resistencia a la vitamina D (relacionada con defectos hereditarios en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la vía metabólica de la vitamina D o en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el receptor Receptor Receptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell. Receptors del calcitriol Calcitriol The physiologically active form of vitamin d. It is formed primarily in the kidney by enzymatic hydroxylation of 25-hydroxycholecalciferol (calcifediol). Its production is stimulated by low blood calcium levels and parathyroid hormone. Calcitriol increases intestinal absorption of calcium and phosphorus, and in concert with parathyroid hormone increases bone resorption. Parathyroid Glands: Anatomy). Las presentaciones clínicas típicas ocurren durante la infancia y se manifiestan como baja estatura, genu valgo o genu varo, dolor Dolor Inflammation óseo y dolor Dolor Inflammation dental. El diagnóstico se realiza mediante estudios de laboratorio y se confirma con la identificación de la mutación en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el gen PHEX PHEX A membrane-bound metalloendopeptidase that may play a role in the degradation or activation of a variety of peptide hormones and intercellular signaling peptides and proteins. Genetic mutations that result in loss of function of this protein are a cause of hypophosphatemic rickets, X-linked dominant. X-linked Hypophosphatemic Rickets. El tratamiento incluye burosumab o dosis altas de vitamina D activada ( calcitriol Calcitriol The physiologically active form of vitamin d. It is formed primarily in the kidney by enzymatic hydroxylation of 25-hydroxycholecalciferol (calcifediol). Its production is stimulated by low blood calcium levels and parathyroid hormone. Calcitriol increases intestinal absorption of calcium and phosphorus, and in concert with parathyroid hormone increases bone resorption. Parathyroid Glands: Anatomy) y fosfato. El monitoreo del paciente por parte de un equipo multidisciplinario es crucial para garantizar el crecimiento adecuado.
Last updated: Dec 15, 2025
El raquitismo hipofosfatémico ligado al AL Amyloidosis cromosoma X (XLHR, por sus siglas en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum inglés) es un trastorno genético raro que causa hipofosfatemia y raquitismo clínico resultante.
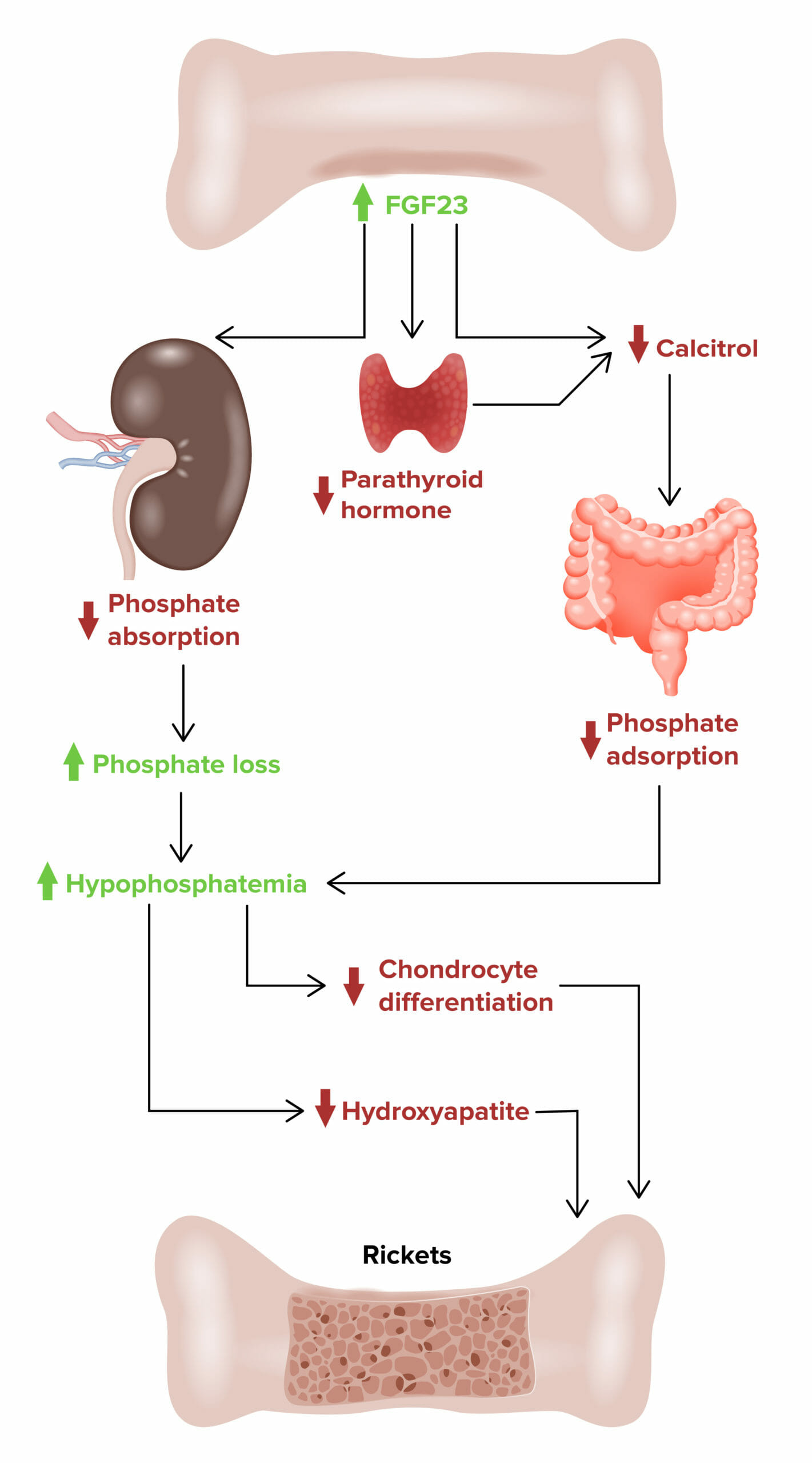
Raquitismo hipofosfatémico ligado al X:
Efecto del factor de crecimiento de fibroblastos 23 (FGF23, por sus siglas en inglés) en el desarrollo del raquitismo hipofosfatémico ligado al cromosoma X
La presentación clínica del XLHR puede variar de asintomática a gravemente sintomática. El raquitismo hipofosfatémico ligado al AL Amyloidosis cromosoma X suele confundirse con el raquitismo nutricional en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria bebés, y los LOS Neisseria síntomas incluyen:
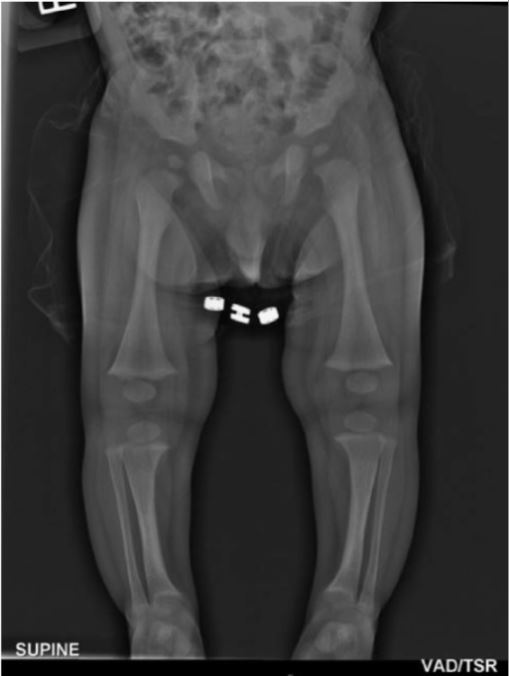
Un lactante de 7 meses con raquitismo hipofosfatémico ligado al cromosoma X:
Los huesos largos de las extremidades inferiores muestran un ensanchamiento de las metáfisis con aspecto deteriorado, pero sin ampliación de la epífisis. También hay un leve arqueo de la tibia.
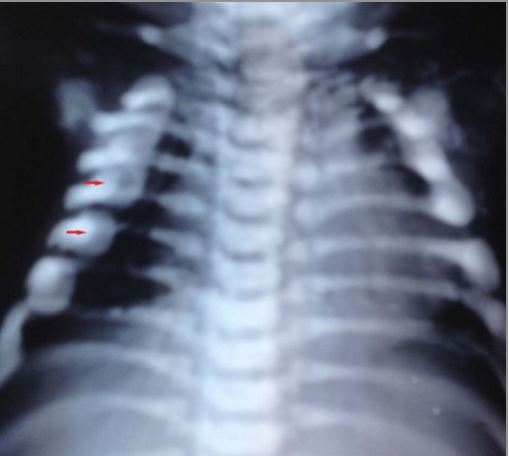
Radiografía que muestra el rosario raquítico
Imagen: “Malignant infantile osteopetrosis: Case report with review of literature” por Essabar L, Meskini T, Ettair S, Erreimi N, Mouane N. Licencia: CC BY 2.0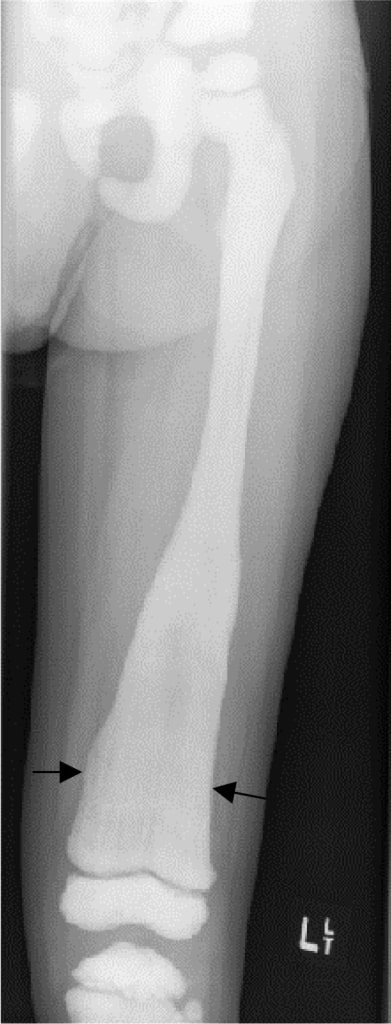
Antecedentes familiares:
Pruebas de laboratorio:
Pruebas genéticas:
Imagenología:
Debe realizarse una evaluación radiológica para descartar arqueo fisiológico y displasia ósea. Los LOS Neisseria posibles hallazgos radiográficos pueden ser:
Los LOS Neisseria objetivos del tratamiento son mejorar la osteomalacia Osteomalacia Disorder caused by an interruption of the mineralization of organic bone matrix leading to bone softening, bone pain, and weakness. It is the adult form of rickets resulting from disruption of vitamin d; phosphorus; or calcium homeostasis. Osteomalacia and Rickets y las deformidades del esqueleto, mejorar el crecimiento de los LOS Neisseria bebés y la actividad física de los LOS Neisseria niños, así como disminuir el dolor Dolor Inflammation.
Algunos adultos con XLHR podrían tener problemas médicos mínimos, mientras que otros pueden experimentar molestias persistentes o complicaciones. Es imprescindible monitorear el tratamiento para detectar posibles complicaciones del hiperparatiroidismo y la nefrocalcinosis.