


A polipose adenomatosa familiar (PAF) é uma doença genética hereditária autossómica dominante que se apresenta com numerosos pólipos adenomatosos no cólon. A polipose adenomatosa familiar é a mais MAIS Androgen Insensitivity Syndrome comum das síndromes de polipose, sendo estas um grupo de doenças hereditárias ou adquiridas caracterizadas pelo crescimento de pólipos no trato GI, associado a outras características extracólicas Estas síndromes são causadas por mutações em genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure específicos associados à supressão tumoral ou regulação do ciclo celular. Todos os doentes com PAF desenvolvem cancro do cólon aos 35 – 40 anos se não forem tratados. O tratamento é feito através de um programa de vigilância e colectomia.
Last updated: Dec 26, 2025
A polipose adenomatosa familiar (PAF) é uma doença autossómica dominante associada ao desenvolvimento de numerosos adenomas colorretais.
Há mutações patogénicas no gene supressor tumoral “adenomatous polyposis coli” ( APC APC A polyposis syndrome due to an autosomal dominant mutation of the apc genes on chromosome 5. The syndrome is characterized by the development of hundreds of adenomatous polyps in the colon and rectum of affected individuals by early adulthood. Familial Adenomatous Polyposis) na banda 5q21 do cromossoma 5:

Resposta normal ao stess e dano celular
Imagem por Lecturio. Licença: CC BY-NC-SA 4.0Existem 4 síndromes da mutação germinativa no gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics APC APC A polyposis syndrome due to an autosomal dominant mutation of the apc genes on chromosome 5. The syndrome is characterized by the development of hundreds of adenomatous polyps in the colon and rectum of affected individuals by early adulthood. Familial Adenomatous Polyposis :
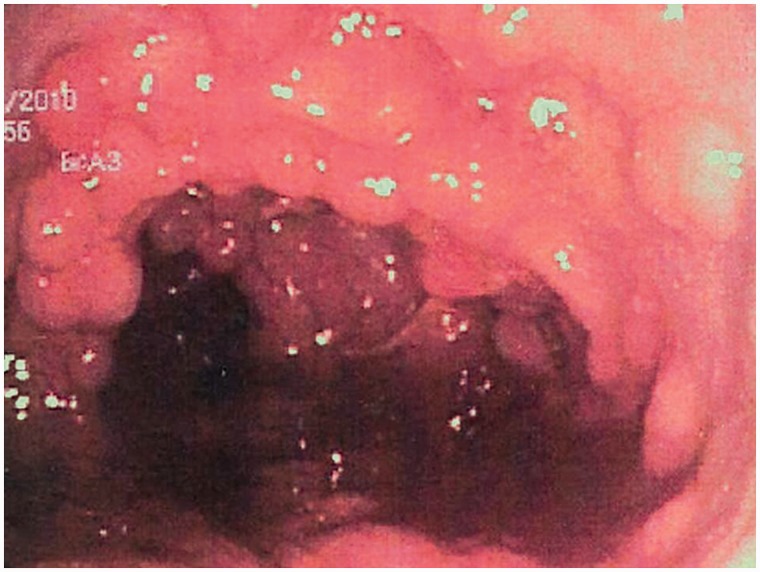
Mucosa do cólon coberta por pólipos adenomatosos na polipose adenomatosa familiar
Imagem : “Colonic mucosa carpeted with adenomatous polyps in a patient with familial adenomatous polyposis” por Shussman, N., Wexner, S.D. Licença: CC BY 3.0
Visão endoscópica de múltiplos adenomas estabelecidos na FAP
Imagem : “Familial adenomatous polyposis” por Half, E., Bercovich, D., Rozen, P. Licença: CC BY 2.0, recortada por Lecturio.