


Os pólipos do cólon são projeções de tecido mucoso no cólon, o local mais MAIS Androgen Insensitivity Syndrome comum de pólipos no trato GI. Os pólipos podem ser classificados como neoplásicos ou não neoplásicos e podem estar associados a síndromes genéticas. Os pólipos hiperplásicos não são neoplásicos e são o tipo mais MAIS Androgen Insensitivity Syndrome comum em geral, enquanto os adenomas são o tipo mais MAIS Androgen Insensitivity Syndrome comum de pólipo neoplásico e têm potencial para evoluir para cancro. Para a maioria das pessoas sem síndromes hereditárias, o rastreamento do cancro de cólon deve começar aos 45 anos e na adolescência para aqueles com a rara síndrome de polipose adenomatosa familiar (PAF). O diagnóstico é por biópsia, e o tratamento inclui vigilância frequente em pessoas com pólipos adenomatosos ou rastreio a cada 10 anos na população geral até os 75 anos.
Last updated: Jan 15, 2026
Cada síndrome ocorre em < 1% da população e é responsável por 5%–10% dos CCRs.
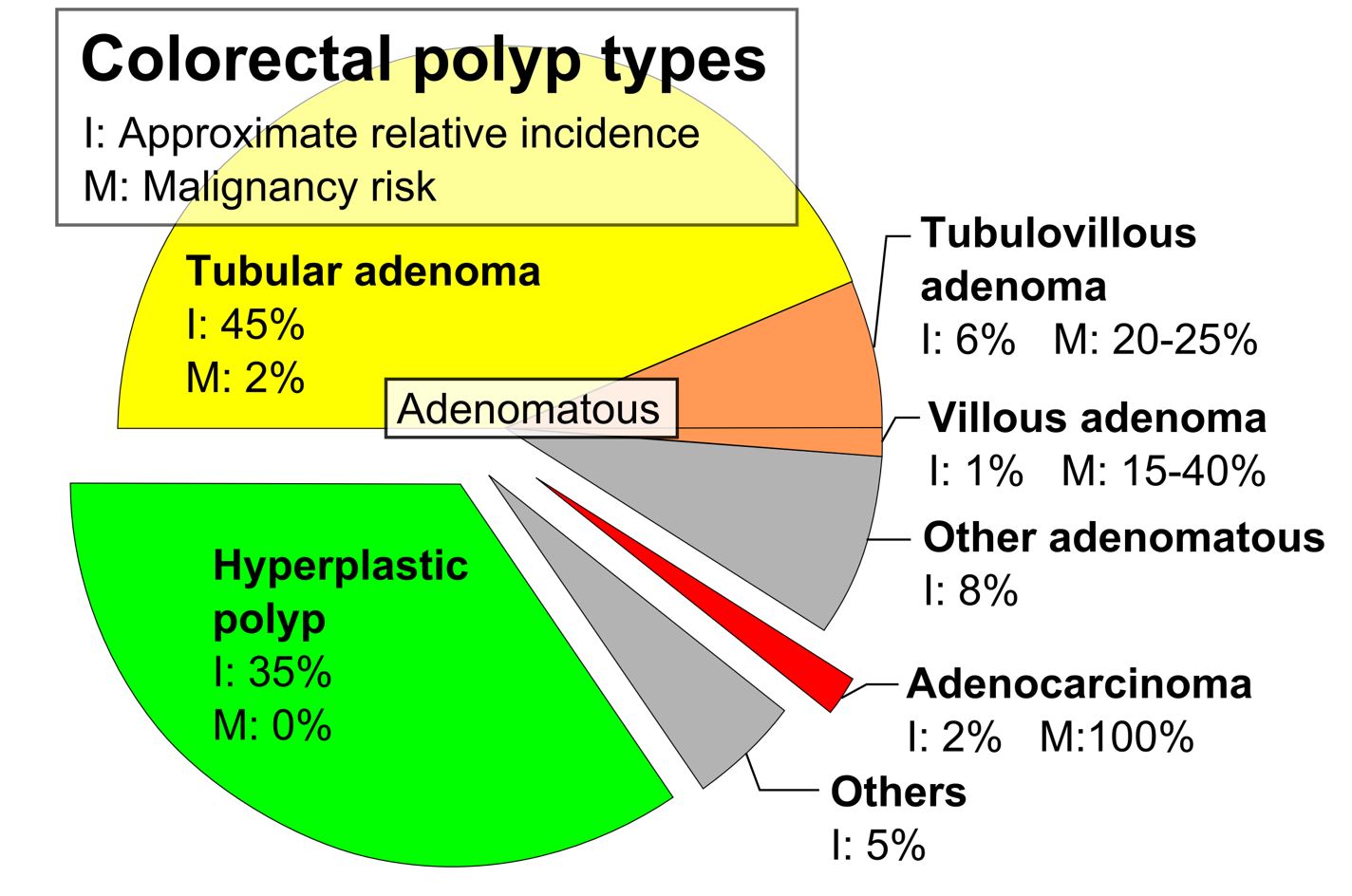
Tipos de pólipos colorretais, incidência relativa aproximada (I) e risco de malignidade (M) (2019)
Imagem: “Pie chart of colorectal polyp etiologies” por Mikael Häggström. Licença: CC0 1.0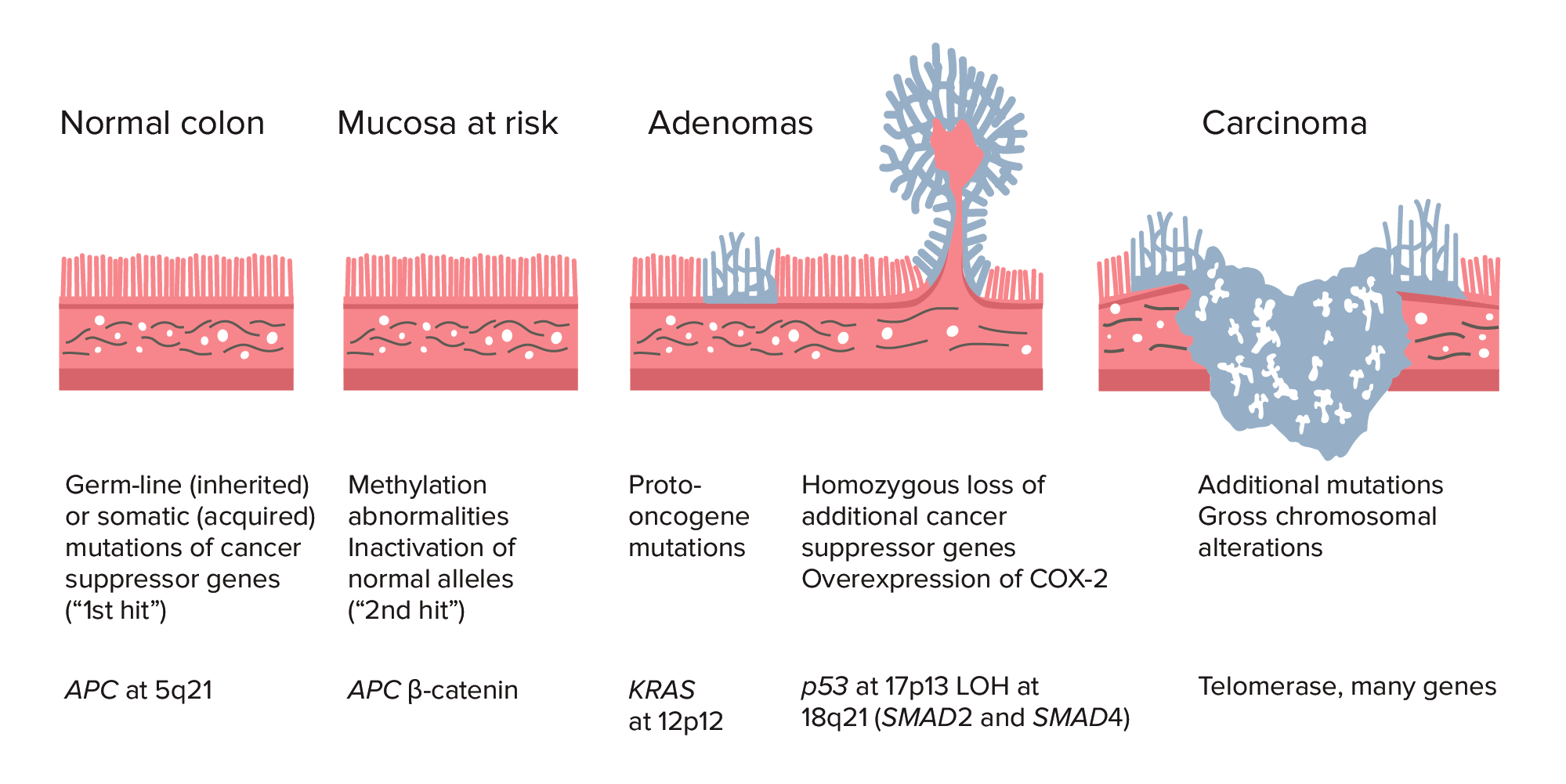
Sequência adenoma-carcinoma do cólon normal ao carcinoma:
A formação do cacnro colorretal (CCR) começa com a mutação do gene APC (congénito ou adquirido) e anomalias da metilação. Outras alterações podem incluir a mutação do gene KRAS. No final do processo, a deleção de p53, perda de heterozigotia (LOH) em 18q21 (SMAD2 e SMAD4), com sobreexpressão de COX-2 pode contribuir para um maior crescimento e progressão para cancro. A acumulação de mutações, ao invés do momento de ocorrência, é mais crucial na carcinogénese.
A maioria dos pólipos do cólon são assintomáticos e descobertos por rastreio de rotina do cancro de cólon.
O diagnóstico definitivo e o tratamento dos pólipos do cólon são alcançados pela remoção endoscópica de todo o pólipo.
Pólipo hiperplásico do reto
Imagem: “Hyperplastic Polyp of the Rectum (14060044206)” por Ed Uthman. Licença: CC BY 2.0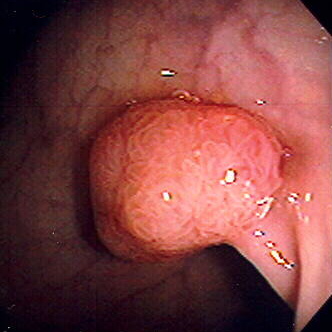
Pólipo do cólon com haste (pedunculado)
Imagem: “Polyp” por Stephen Holland. Licença: CC BY 2.5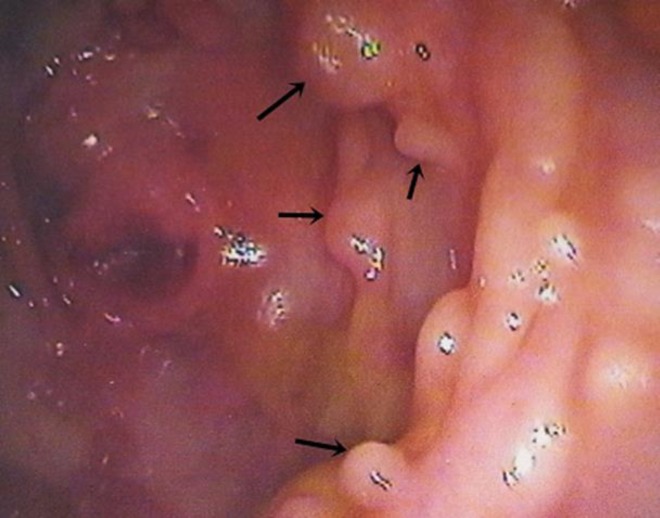
Múltiplos pólipos no cólon, variando de 0,5 a 1 cm de diâmetro
Imagem: “Multiple polyps in colon” por Agrawal N, Santra T, Kar A, Guha P, Bar M, Adhikary A, Datta S. Licença: CC BY 3.0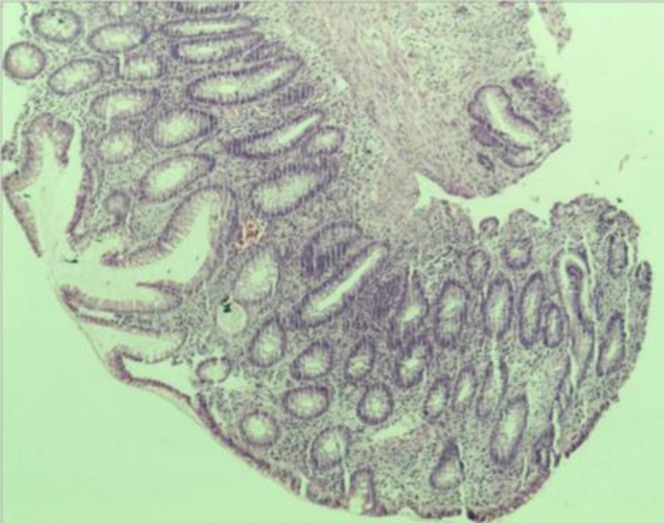
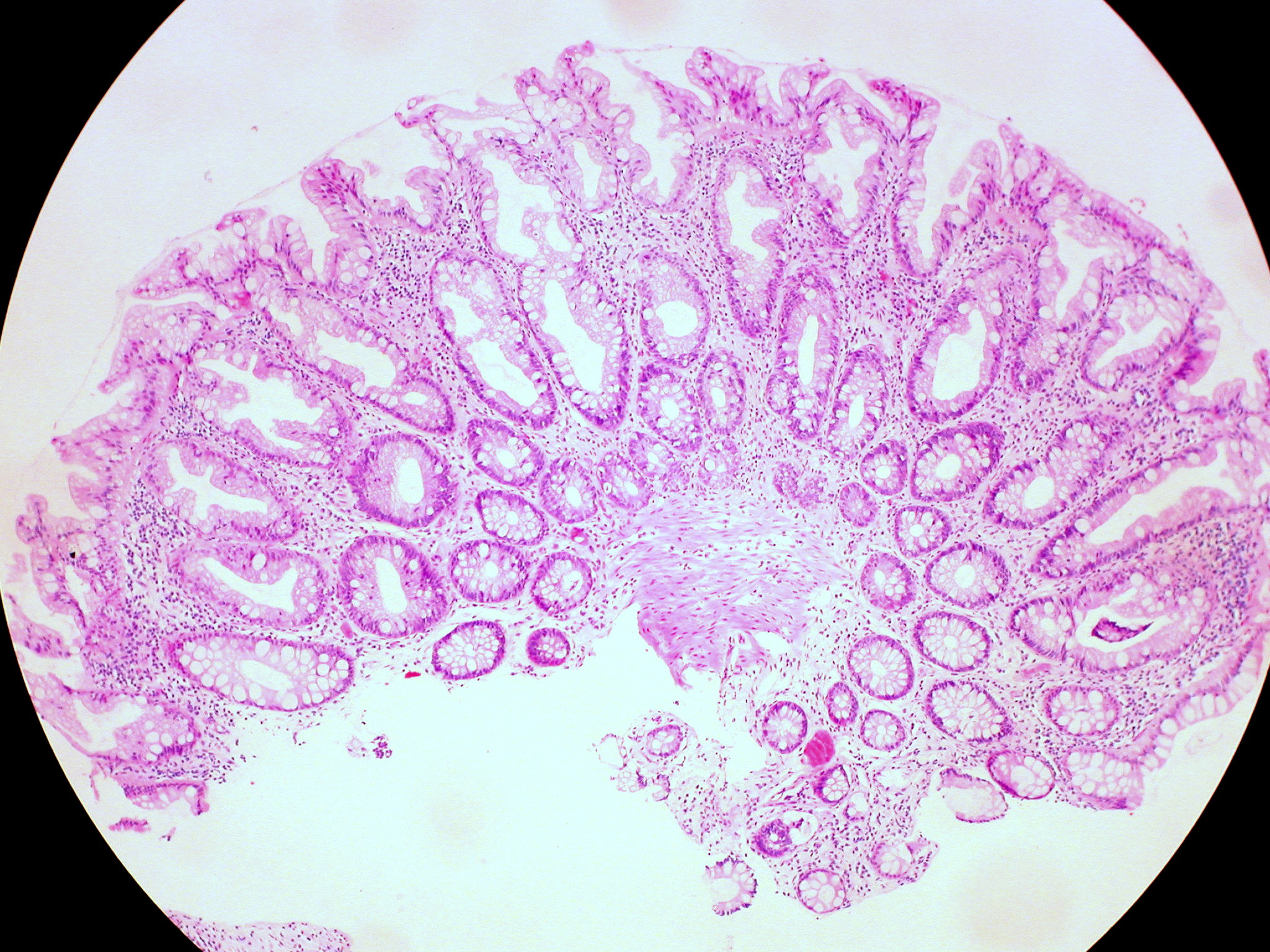
Biópsia do pólipo sigmóide que revela pólipo adenomatoso sem evidência de malignidade
Imagem: “Biopsy from sigmoid polyp revealing adenomatous polyp without any evidence of malignancy” por Agrawal N, Santra T, Kar A, Guha P, Bar M, Adhikary A, Datta S. Licença: CC BY 3.0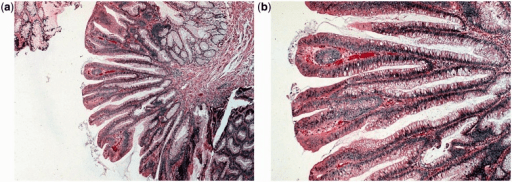
Adenoma viloso: A aparência histológica é de projeções longas em forma de dedos.
a: vista ampliada de baixa potência (coloração H&E, 400x)
b: vista ampliada de alta potência (coloração H&E, 2000x)
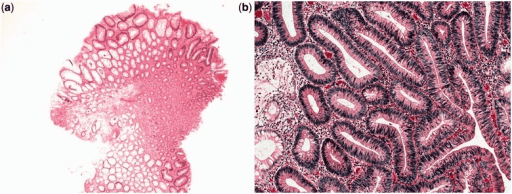
Adenoma tubular: O aspeto histológico é de glândulas tubulares ramificadas.
a: vista ampliada de baixa potência (coloração H&E, 400x)
b: vista ampliada de alta potência (coloração H&E, 2000x)
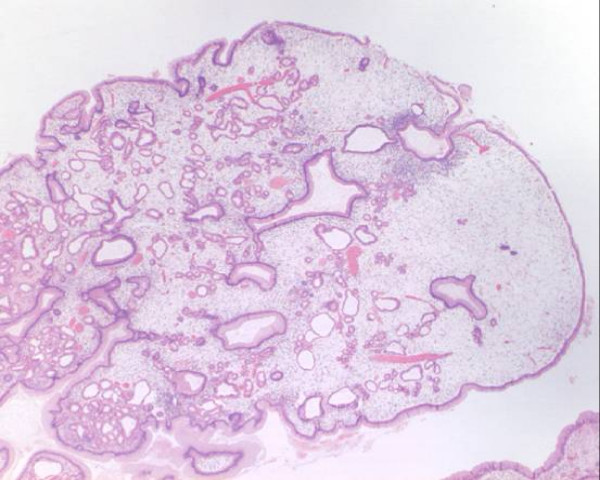
Histologia do pólipo juvenil que mostra glândulas cisticamente dilatadas características:
Mesmo em baixa potência, a natureza benigna e não neoplásica é suportada pela visualização de apenas 1 camada de células epiteliais na superfície e revestindo os espaços glandulares.
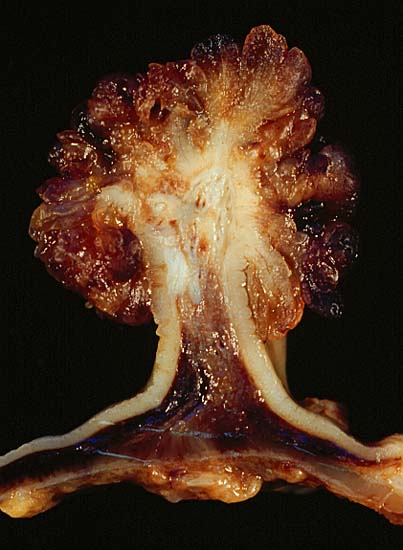
Pólipo Tubuloviloso
Imagem: “Tubulovillous Polyp of the Colon 2” por Ed Uthman. Licença: Domínio público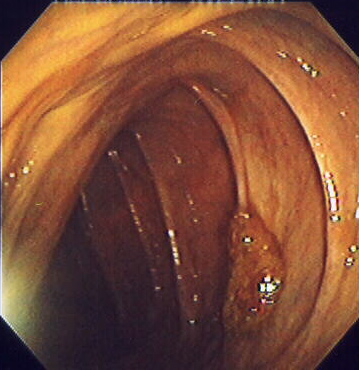
Pólipo do cólon séssil visto em colonoscopia
Imagem: “Endomucosal resection 1” por Stephen Holland. Licença: 2.5Orientações de follow-up após colonoscopia e polipectomia (com base no 2020 Consensus Update by the U.S. Multi-Society Task Force on Colorectal Cancer Colorectal cancer Colorectal cancer (CRC) is the 2nd leading cause of cancer-related deaths in the United States. Colorectal cancer is a heterogeneous disease that arises from genetic and epigenetic abnormalities, with influence from environmental factors. Colorectal Cancer):
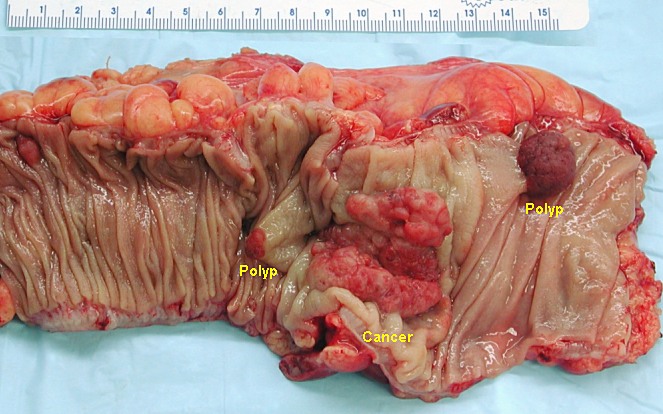
Segmento de cólon ressecado no estado não fixo que contém um carcinoma colorretal invasivo e 2 pólipos adenomatosos
Imagem: “Colon cancer” por Emmanuelm. Licença: CC BY 3.0