


O neuroblastoma Neuroblastoma Neuroblastoma is a malignancy that arises from the neural crest cell derivatives along the sympathetic chain (neuroblasts) and is most commonly located in the adrenal medulla. The tumor often presents in childhood with a flank mass that crosses the midline. Neuroblastoma é uma malignidade que surge dos derivados das células da crista neural ao longo da cadeia simpática (neuroblastos) e é mais MAIS Androgen Insensitivity Syndrome comumente localizado na medula adrenal. O tumor Tumor Inflammation geralmente se apresenta na infância com uma massa no flanco que cruza a linha média. O neuroblastoma Neuroblastoma Neuroblastoma is a malignancy that arises from the neural crest cell derivatives along the sympathetic chain (neuroblasts) and is most commonly located in the adrenal medulla. The tumor often presents in childhood with a flank mass that crosses the midline. Neuroblastoma também pode se manifestar como síndrome paraneoplásica opsoclonia-mioclonia. O tumor Tumor Inflammation é diagnosticado através de biópsia, e os dados de apoio incluem a medição dos produtos de degradação das catecolaminas, como ácido vanilmandélico (VMA) e ácido homovanílico (HVA) na urina. Estudos de imagem são necessários para localizar o tumor Tumor Inflammation. O manejo depende de vários fatores, como o estágio da malignidade e a idade do paciente no momento do diagnóstico. O prognóstico é favorável nos estágios iniciais do neuroblastoma Neuroblastoma Neuroblastoma is a malignancy that arises from the neural crest cell derivatives along the sympathetic chain (neuroblasts) and is most commonly located in the adrenal medulla. The tumor often presents in childhood with a flank mass that crosses the midline. Neuroblastoma.
Last updated: Dec 15, 2025
O neuroblastoma Neuroblastoma Neuroblastoma is a malignancy that arises from the neural crest cell derivatives along the sympathetic chain (neuroblasts) and is most commonly located in the adrenal medulla. The tumor often presents in childhood with a flank mass that crosses the midline. Neuroblastoma, um tumor Tumor Inflammation sólido maligno extracraniano, é uma neoplasia neuroendócrina originada de células-tronco do sistema nervoso simpático (neuroblastos).
| Categorias | Tumores específicos |
|---|---|
| Tumores neuroepiteliais no SNC |
|
| Tumores meníngeos |
|
| Tumores da região selar |
|
| Linfoma primário do SNC | Linfoma primário do SNC |
| Metástase para o cérebro (5x mais MAIS Androgen Insensitivity Syndrome comum do que tumores cerebrais primários) | Mais MAIS Androgen Insensitivity Syndrome comumente decorrentes de: |
| Tumores periféricos |
|
Os neuroblastomas desenvolvem-se a partir de mutações em células primitivas dos gânglios simpáticos (que incluem a medula suprarrenal). As células são conhecidas como neuroblastos e derivam das células da crista neural.
A suspeita diagóstica baseia-se na história e nos achados do exame objetivo. Os exames laboratoriais chave para os neuroblastomas mostram evidência de elevação das catecolaminas. A imagiologia é usada para identificar o tumor Tumor Inflammation, e é necessário biópsia para o diagnóstico definitivo.
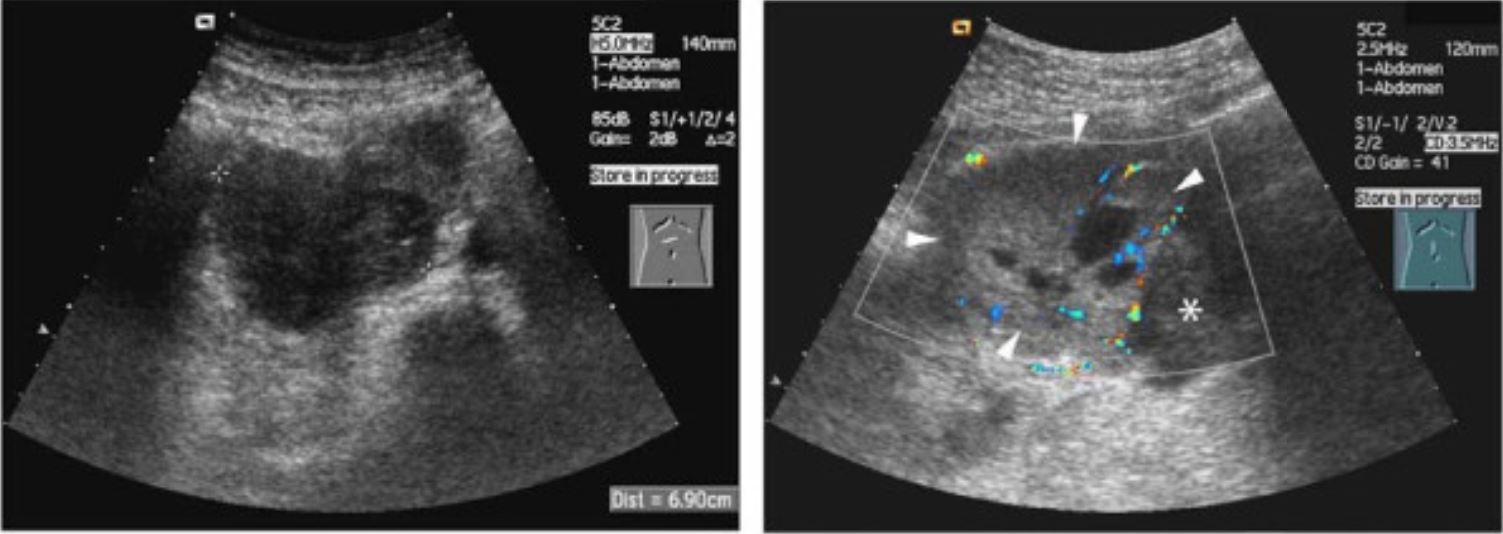
Ultrassonografia de neuroblastoma:
A aparência clássica é uma massa heterogênea com vascularização interna.
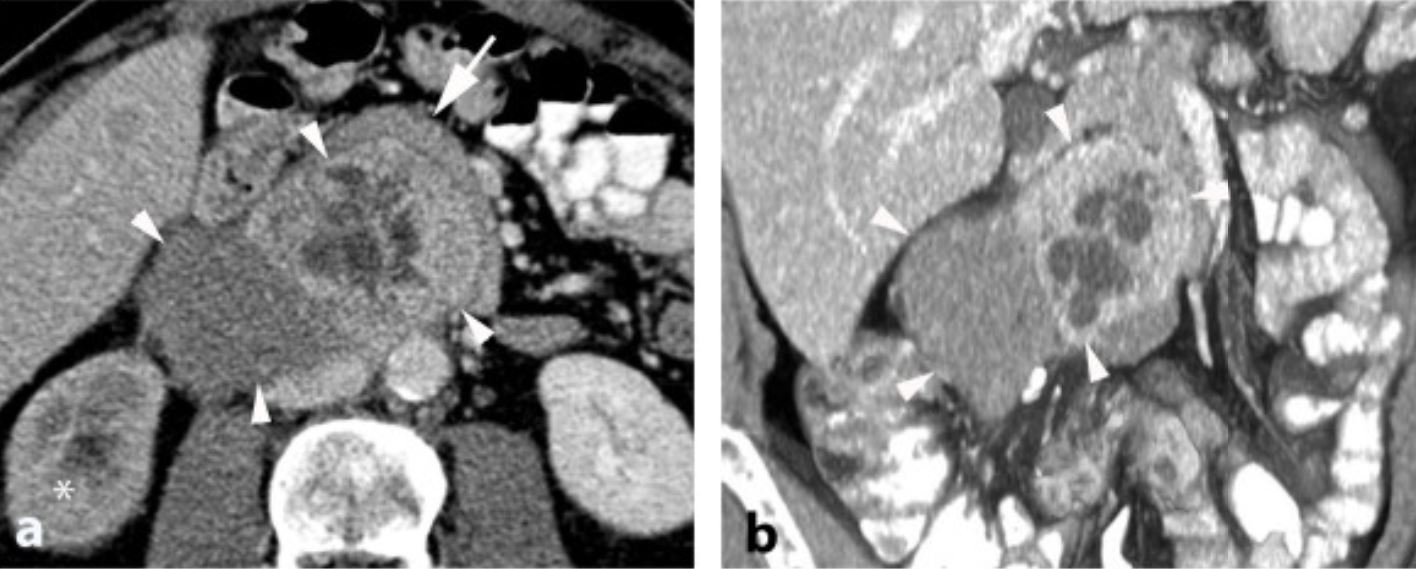
TC de neuroblastoma:
O tumor geralmente aparece heterogêneo com calcificações.
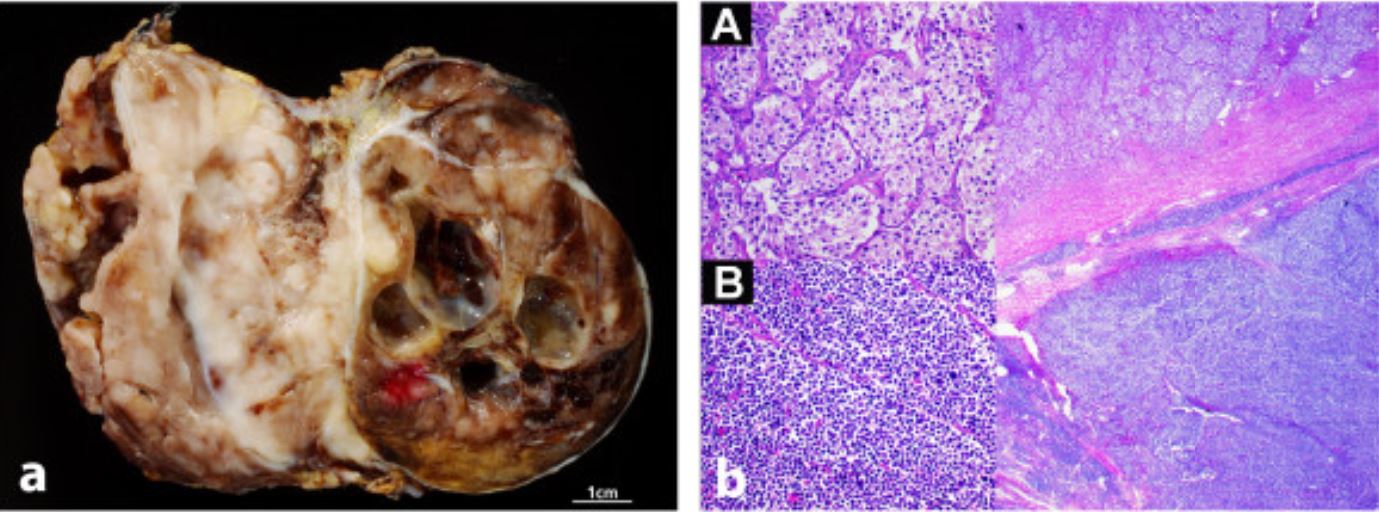
Neuroblastoma na aparência macroscópica e histológica: imagens macroscópicas (esquerda) e microscópicas (direita) de neuroblastoma
Imagem: “Achados radiológicos e patológicos de um paraganglioma composto metastático com neuroblastoma em um homem: relato de caso” por Fritzsche FR, Bode PK, Koch S, Frauenfelder T. Licença: CC BY 2.0Existem múltiplos sistemas de estadiamento, mas o INSS é um dos mais MAIS Androgen Insensitivity Syndrome comuns, São necessários imagiologia do corpo, uma biópsia do tumor Tumor Inflammation primário, biópsia da medula óssea e scan ósseo:
O manejo do neuroblastoma Neuroblastoma Neuroblastoma is a malignancy that arises from the neural crest cell derivatives along the sympathetic chain (neuroblasts) and is most commonly located in the adrenal medulla. The tumor often presents in childhood with a flank mass that crosses the midline. Neuroblastoma depende se o paciente é considerado um neuroblastoma Neuroblastoma Neuroblastoma is a malignancy that arises from the neural crest cell derivatives along the sympathetic chain (neuroblasts) and is most commonly located in the adrenal medulla. The tumor often presents in childhood with a flank mass that crosses the midline. Neuroblastoma de baixo, intermediário ou alto risco. A doença localizada é muitas vezes curável.
A estratificação de risco depende do estágio da malignidade no momento do diagnóstico, amplificação MYCN, deleção do cromossomo 1p e idade no momento do diagnóstico (idade tardia do diagnóstico = pior prognóstico).
