El neuroblastoma Neuroblastoma Neuroblastoma is a malignancy that arises from the neural crest cell derivatives along the sympathetic chain (neuroblasts) and is most commonly located in the adrenal medulla. The tumor often presents in childhood with a flank mass that crosses the midline. Neuroblastoma es una neoplasia que surge de las células de la cresta neural que se deriva a lo largo de la cadena simpática (neuroblastos) y se localiza con mayor frecuencia en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la médula suprarrenal. El tumor Tumor Inflammation suele presentarse en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la infancia con una masa en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el flanco que cruza la línea media. El neuroblastoma Neuroblastoma Neuroblastoma is a malignancy that arises from the neural crest cell derivatives along the sympathetic chain (neuroblasts) and is most commonly located in the adrenal medulla. The tumor often presents in childhood with a flank mass that crosses the midline. Neuroblastoma también puede manifestarse como un síndrome paraneoplásico opsoclono-mioclono. El tumor Tumor Inflammation se diagnostica mediante biopsia y los LOS Neisseria datos de soporte incluyen la medición de los LOS Neisseria productos de descomposición de las catecolaminas, como el ácido vanilmandélico y el ácido homovanílico en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la orina. Los LOS Neisseria estudios de imagenología son necesarios para localizar el tumor Tumor Inflammation. El tratamiento depende de varios factores, como el estadio de la malignidad y la edad del paciente en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el momento del diagnóstico. El pronóstico es favorable en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum las primeras fases del neuroblastoma Neuroblastoma Neuroblastoma is a malignancy that arises from the neural crest cell derivatives along the sympathetic chain (neuroblasts) and is most commonly located in the adrenal medulla. The tumor often presents in childhood with a flank mass that crosses the midline. Neuroblastoma.
Last updated: Dec 15, 2025
El neuroblastoma Neuroblastoma Neuroblastoma is a malignancy that arises from the neural crest cell derivatives along the sympathetic chain (neuroblasts) and is most commonly located in the adrenal medulla. The tumor often presents in childhood with a flank mass that crosses the midline. Neuroblastoma, un tumor Tumor Inflammation sólido maligno extracraneal, es una neoplasia neuroendocrina que se origina en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum las células madre del sistema nervioso simpático (neuroblastos).
| Categorías | Tumores específicos |
|---|---|
| Tumores neuroepiteliales en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el sistema nervioso central (SNC) |
|
| Tumores meníngeos |
|
| Tumores de la región selar |
|
| Linfoma primario del SNC | Linfoma primario del SNC |
| Metástasis al AL Amyloidosis cerebro (5 veces más común que los LOS Neisseria tumores cerebrales primarios) | Más comúnmente surgen de: |
| Tumores periféricos |
|
Los LOS Neisseria neuroblastomas surgen de mutaciones en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum las células primitivas del ganglio simpático (que incluye la médula suprarrenal). Las células se conocen como neuroblastos y derivan de las células de la cresta neural.
El diagnóstico se sospecha sobre la base de los LOS Neisseria antecedentes y los LOS Neisseria hallazgos del examen físico. Las pruebas de laboratorio clave para los LOS Neisseria neuroblastomas muestran evidencia de catecolaminas elevadas. Los LOS Neisseria estudios de imagen se utiliza para identificar el tumor Tumor Inflammation, y se requiere una biopsia para el diagnóstico definitivo.
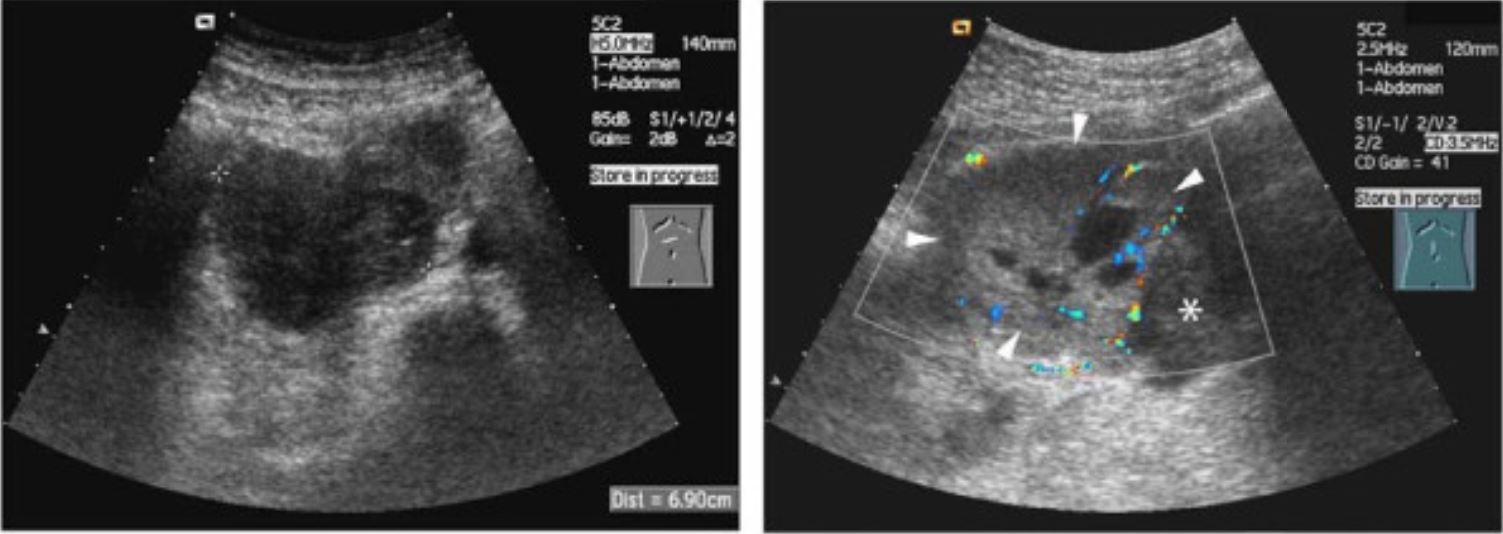
Ultrasonido del neuroblastoma:
El aspecto clásico es una masa heterogénea con irrigación interna.
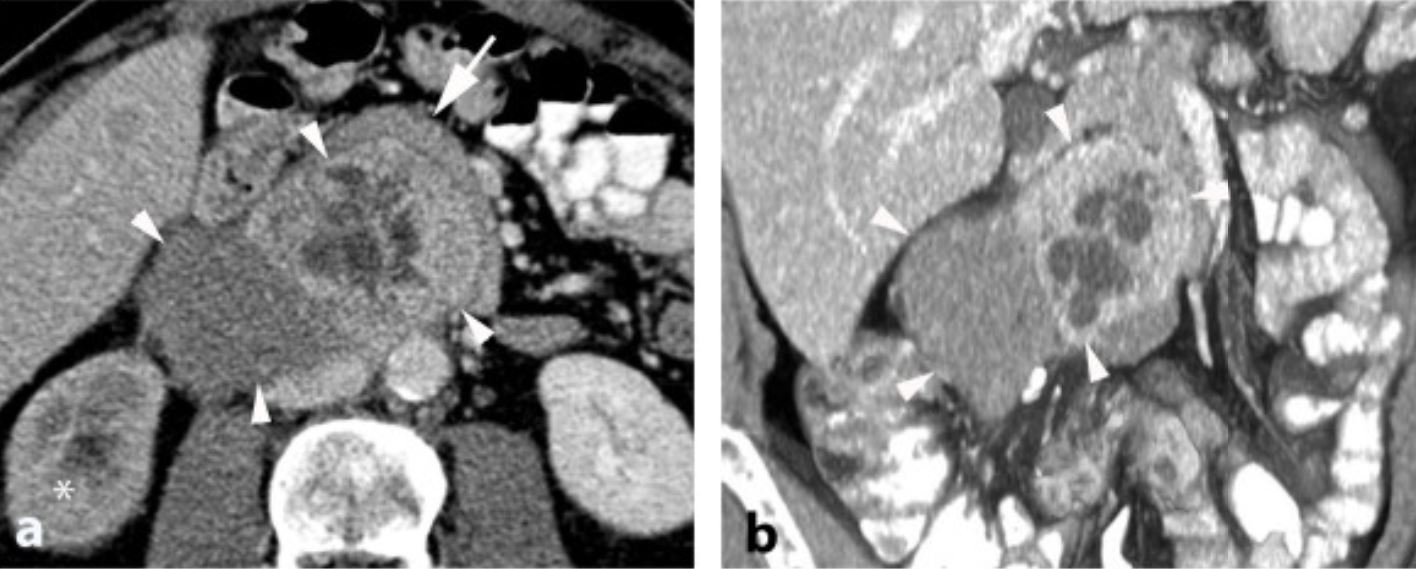
TC del neuroblastoma:
El tumor suele tener un aspecto heterogéneo con calcificaciones.
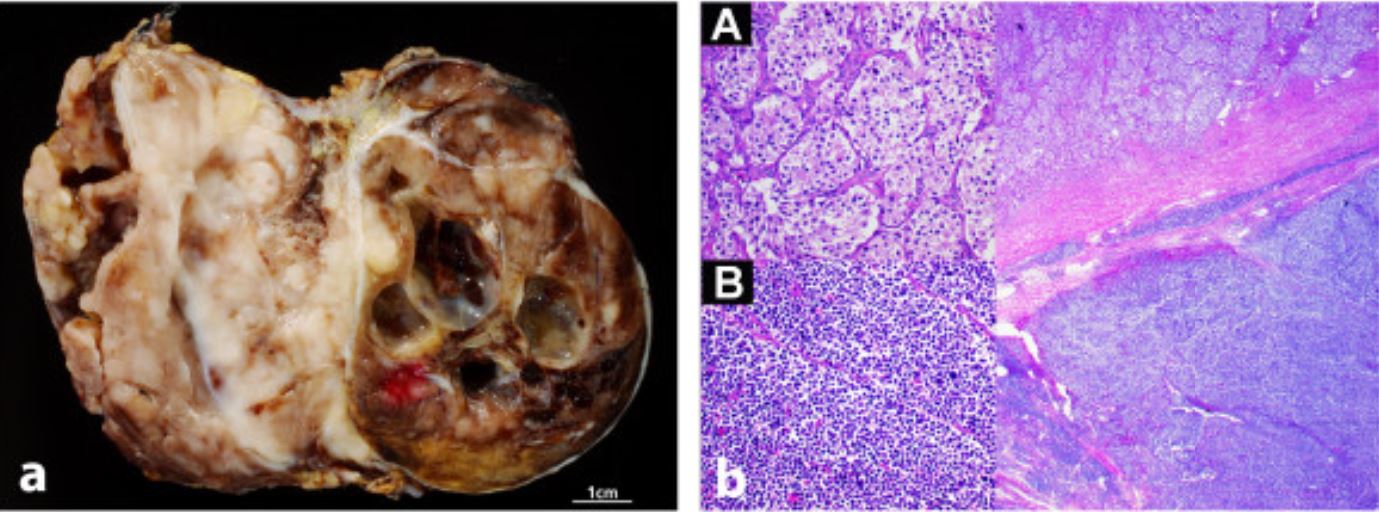
Neuroblastoma en aspecto completo e histológico: Imágenes macroscópicas (izquierda) y microscópicas (derecha) del neuroblastoma
Imagen: “Radiological and pathological findings of a metastatic composite paraganglioma with neuroblastoma in a man: a case report” por Fritzsche FR, Bode PK, Koch S, Frauenfelder T. Licencia: CC BY 2.0Existen múltiples sistemas de estadificación, pero el INSS es uno de los LOS Neisseria más comunes. Se requieren imágenes corporales, una biopsia del tumor Tumor Inflammation primario, una biopsia de médula ósea y una gammagrafía ósea:
El tratamiento del neuroblastoma Neuroblastoma Neuroblastoma is a malignancy that arises from the neural crest cell derivatives along the sympathetic chain (neuroblasts) and is most commonly located in the adrenal medulla. The tumor often presents in childhood with a flank mass that crosses the midline. Neuroblastoma depende de si se considera que el paciente tiene un neuroblastoma Neuroblastoma Neuroblastoma is a malignancy that arises from the neural crest cell derivatives along the sympathetic chain (neuroblasts) and is most commonly located in the adrenal medulla. The tumor often presents in childhood with a flank mass that crosses the midline. Neuroblastoma de bajo, intermedio o alto riesgo. La enfermedad localizada suele ser curable.
La estratificación del riesgo depende del estadio de la neoplasia en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el momento del diagnóstico, de la amplificación de NMYC, de la deleción del cromosoma 1p y de la edad en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el momento del diagnóstico (edad más tardía del diagnóstico = peor pronóstico).