


As mucopolissacaridoses, um subconjunto das doenças de depósito lisossómico, são um grupo de doenças hereditárias caracterizadas por ausência ou defeito das enzimas necessárias para degradar as cadeias de hidratos de carbono denominadas glicosaminoglicanos (GAGs), previamente conhecidos como mucopolissacarídeos. Estas doenças levam à acumulação de GAGs dentro dos lisossomas celulares, o que resulta numa variedade de problemas de saúde. A maioria dos doentes parece saudável ao nascimento, mas a função física e/ou mental deteriora à medida que a acumulação progride. Com a progressão da doença, podem ser afetados vários sistemas orgânicos. O diagnóstico pode ser feito pela medição das concentrações de GAG na urina e através de ensaios enzimáticos para identificar a deficiência enzimática. O tratamento depende da doença específica, do grau de acumulação de GAG e do grau de deformidade.
Last updated: Apr 23, 2025
Mucopolissacaridoses ( MPSs MPSs The mucopolysaccharidoses, a subset of the lysosomal storage diseases, are a group of inherited disorders characterized by absent or defective enzymes needed to break down carbohydrate chains called glycosaminoglycans (GAGs). These disorders lead to the accumulation of gags within cells. Mucopolysaccharidoses) são um grupo de doenças metabólicas genéticas devido a enzimas ausentes ou defeituosas necessárias para degradar as cadeias de hidratos de carbono, chamadas glicosaminoglicanos (GAGs).
As MPSs MPSs The mucopolysaccharidoses, a subset of the lysosomal storage diseases, are a group of inherited disorders characterized by absent or defective enzymes needed to break down carbohydrate chains called glycosaminoglycans (GAGs). These disorders lead to the accumulation of gags within cells. Mucopolysaccharidoses são todas herdadas e classificadas com base na deficiência enzimática.
Os indivíduos afetados geralmente não são afetados ao nascimento, mas experimentam progressão da doença à medida que envelhecem. As características clínicas variam de acordo com o tipo de MPS.
Espectro patológico contínuo que se divide em 3 entidades clínicas com base na gravidade da doença (as formas menos graves são referidas como atenuadas):
Características clínicas distintivas ( mais MAIS Androgen Insensitivity Syndrome ou menos pronunciadas dependendo da gravidade):
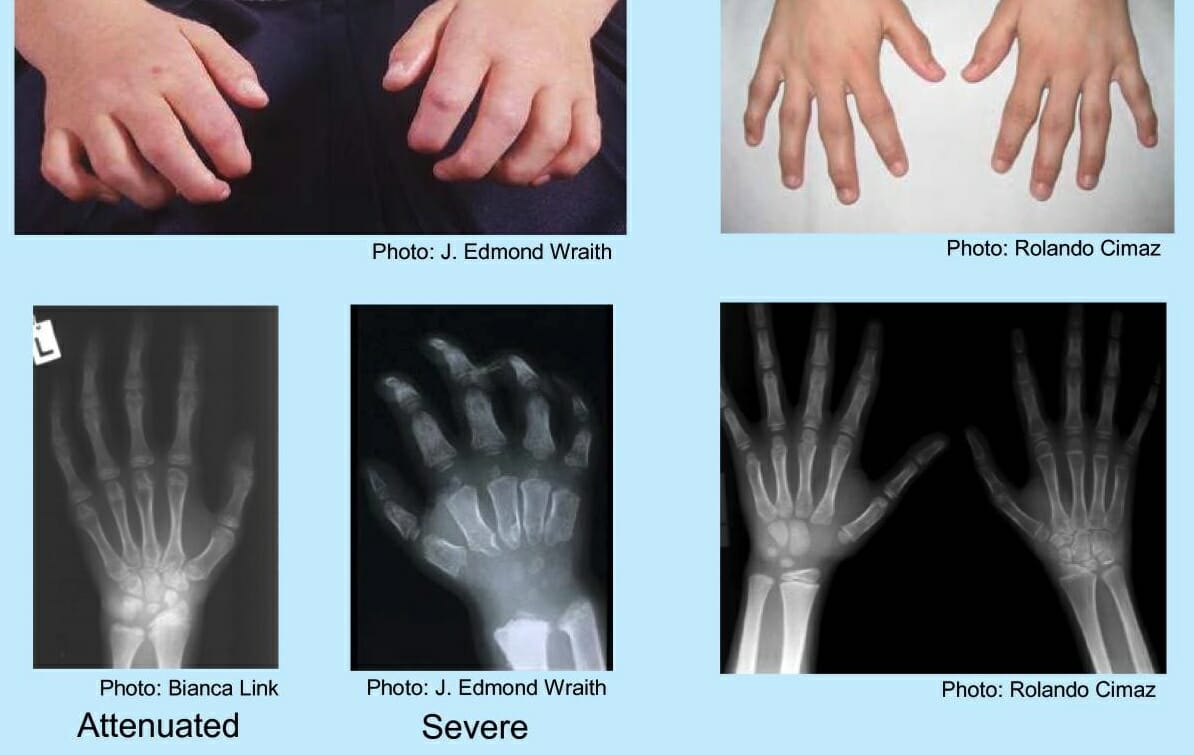
Mãos de uma criança com mucopolissacaridose:
Os achados incluem mão em “garra”, ossos do metacarpo anormais, alargamento proximal das falanges e deformidade em V do rádio distal e cúbito.
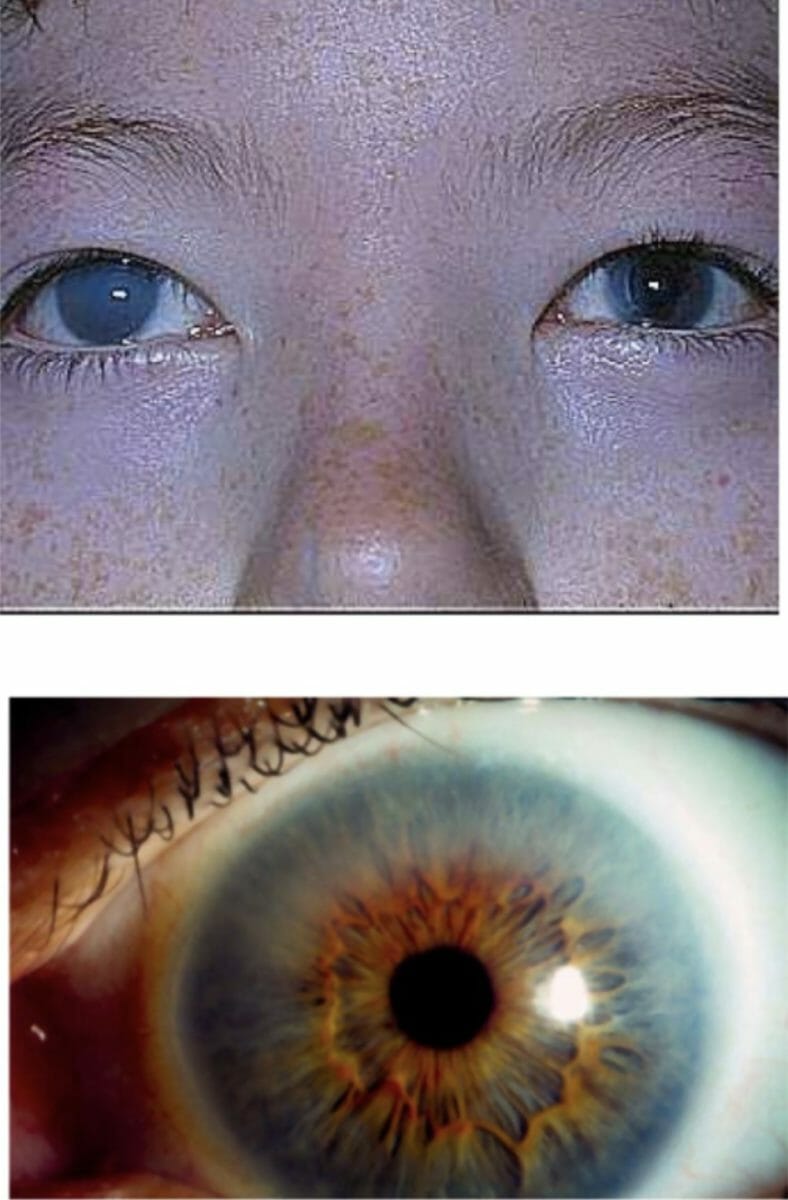
Opacidade da córnea em doente com MPS I: Este achado pode ser grave, como mostra a imagem de cima, ou pode ser menos evidente, como mostra a imagem abaixo.
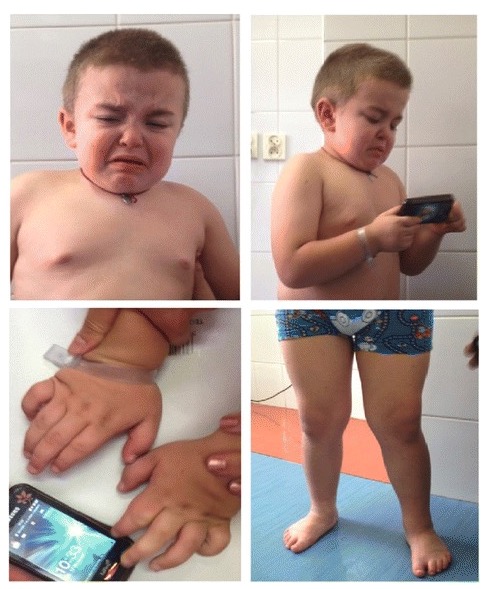
Características clínicas da mucopolissacaridose IIIA:
Dismorfismo facial (características faciais grosseiras, ponte nasal levemente deprimida, sobrancelhas proeminentes, orelhas de implantação baixa, má oclusão, bochechas cheias, cabelos crespos e secos, e pescoço curto) e sintomas esqueléticos (joelhos valgos, pés em varo e mãos robustas).
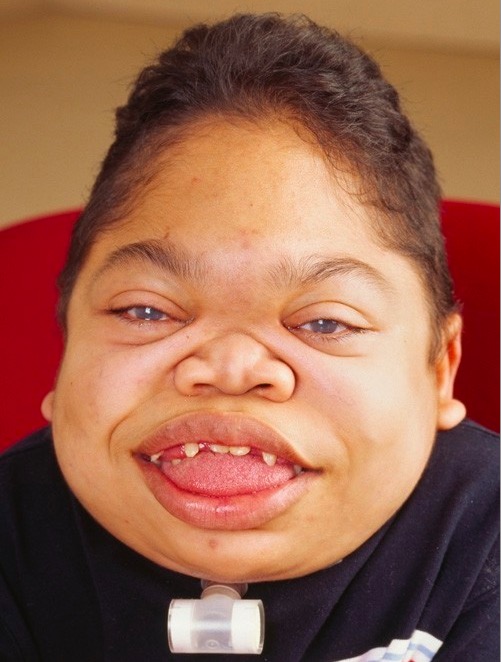
MPS VI de progressão rápida em doente de 16 anos:
A face apresenta fácies grosseira, com bossa frontal, língua aumentada, lábios grossos, dentição anormal e hiperplasia gengival.
História clínica e exame objetivo:
Análise laboratorial:
Não há cura para MPS. A assistência médica visa tratar condições sistémicas e melhorar a qualidade de vida. Atualmente, o eventual declínio na função é inevitável.
