


O grupo heme corresponde a uma porfirina que contem ferro (constituída por 4 grupos pirrol), sintetizada maioritariamente na medula óssea e no fígado. O grupo heme faz parte da constituição de muitas substâncias essenciais, incluindo dos citocromos, da mioglobina e hemoglobina. As suas funções biológicas são, o transporte de gases (por exemplo, O2) e a transferência de eletrões. A biossíntese do grupo heme é um processo constituído por 8 etapas, iniciado pela síntese do ácido aminolevulínico. A disponibilidade do ferro interfere com a produção do grupo heme, dado que a última etapa envolve a inserção do ferro. Este ião é obtido através da dieta e da decomposição de produtos que contém grupo heme. Nos processos catabólicos, o heme é convertido em pigmentos biliares, ocorrendo a produção de bilirrubina. As mutações associadas às enzimas que participam na síntese do grupo heme levam a um grupo de patologias conhecidas como porfirias, e os defeitos no catabolismo do heme causam hiperbilirrubinemias.
Last updated: Apr 25, 2025
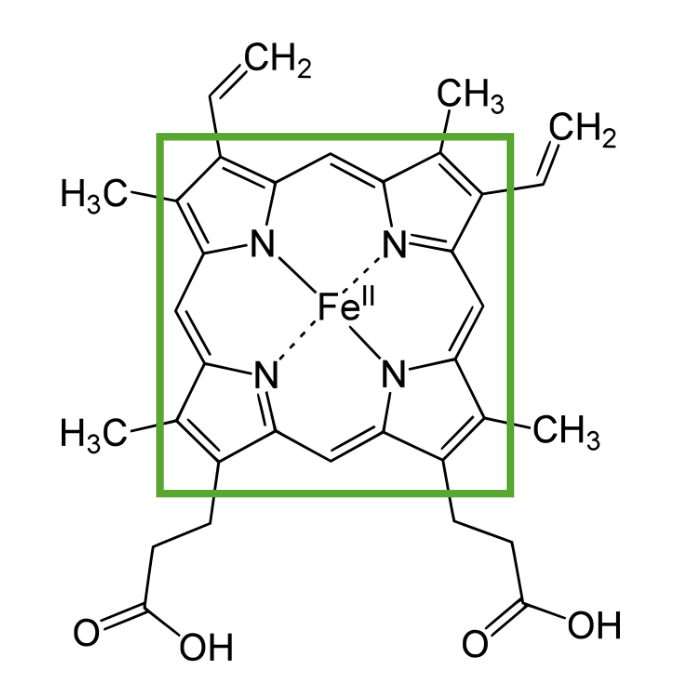
A imagem apresentada representa a estrutura do grupo heme e evidência a presença do anel de porfirina com ião de ferro no centro
Imagem por Lecturio.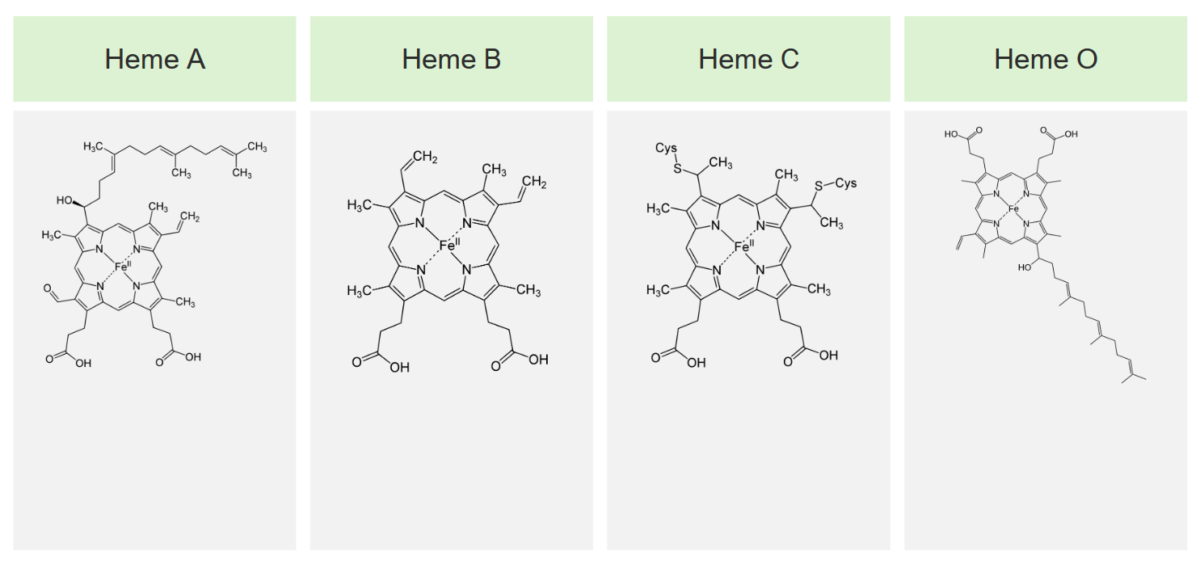
Os 4 principais tipos de grupo heme e as suas estruturas
Imagem por Lecturio.O grupo heme é sintetizado nos normoblastos, mas não nos eritrócitos maduros. A sua biossíntese ocorre em 8 etapas.
A 1.ª etapa consiste na síntese do ácido aminolevulínico.
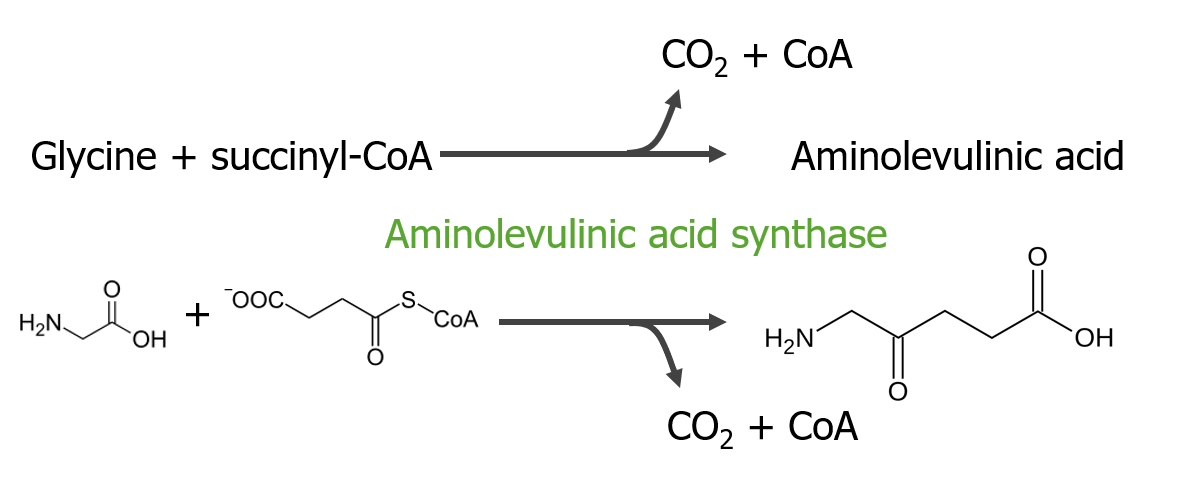
1.ª etapa do metabolismo do grupo heme
Imagem por Lecturio.A 2.ª etapa consiste na formação de porfobilinogénio ( PBG PBG Heme Metabolism).
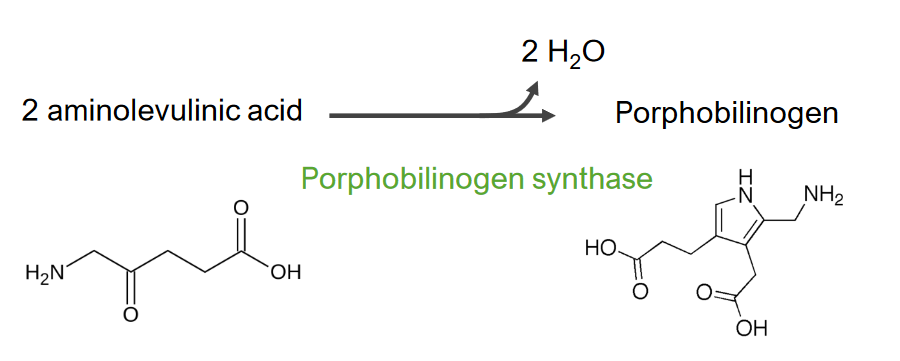
2.ª etapa do metabolismo do grupo heme
Formação de porfobilinogénio
A 3.ª etapa consiste na formação de hidroximetilbilano ( HMB HMB Excessive menstrual blood loss (objectively defined as > 80 mL blood loss/cycle). Can be based on heavy flow, as determined by the patient Abnormal Uterine Bleeding).
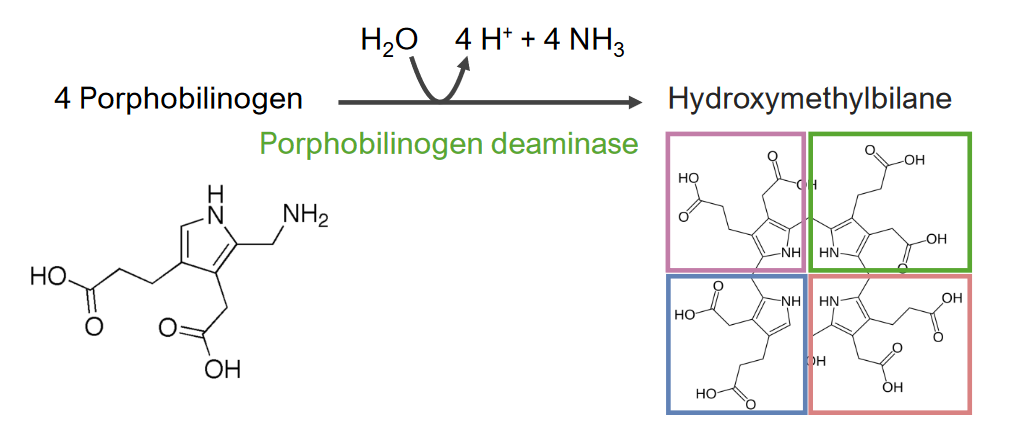
3.ª etapa do metabolismo do grupo heme:
Formação de hidroximetilbilano
A 4.ª etapa consiste na formação de uroporfirinogénio (UPG).
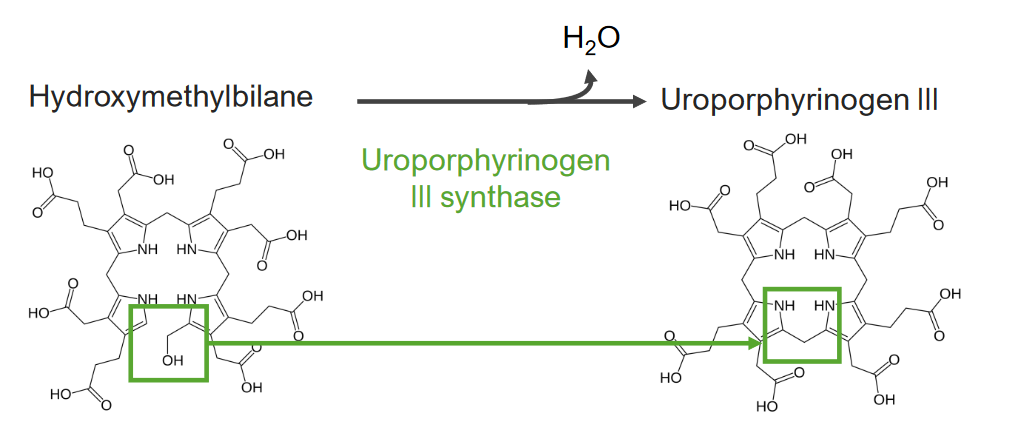
4.ª etapa do metabolismo do grupo heme:
Formação do uroporfirinogénio
A 5.ª etapa consiste na síntese de coproporfirinogénio (CPG) III.
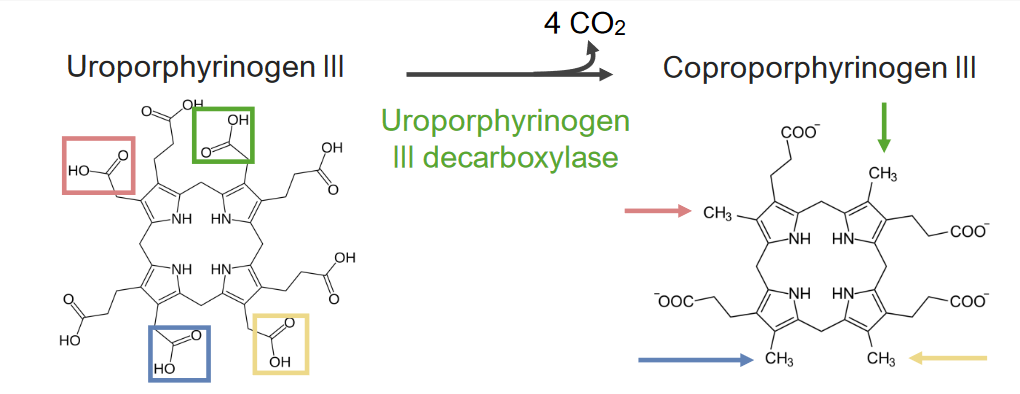
5.ª etapa do metabolismo do grupo heme:
Formação de coproporfirinogénio III
A 6.ª etapa corresponde à síntese de protoporfirinogénio (PPG).
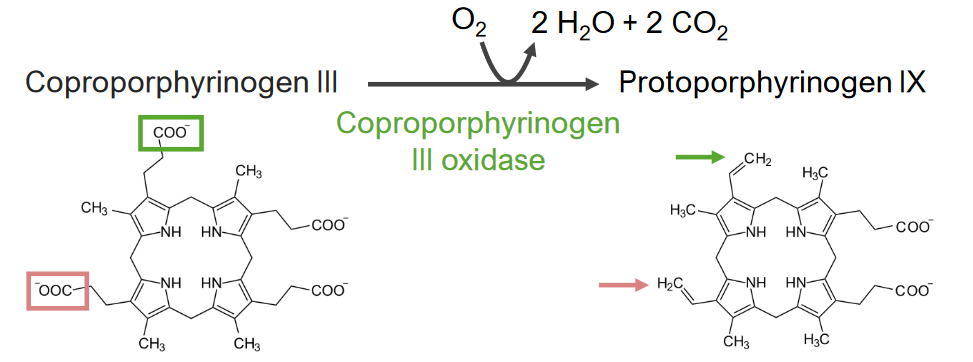
6.ª etapa do metabolismo do grupo heme:
Síntese de protoporfirinogêéio
A 7.ª etapa consiste na formação de protoporfirina (PP).
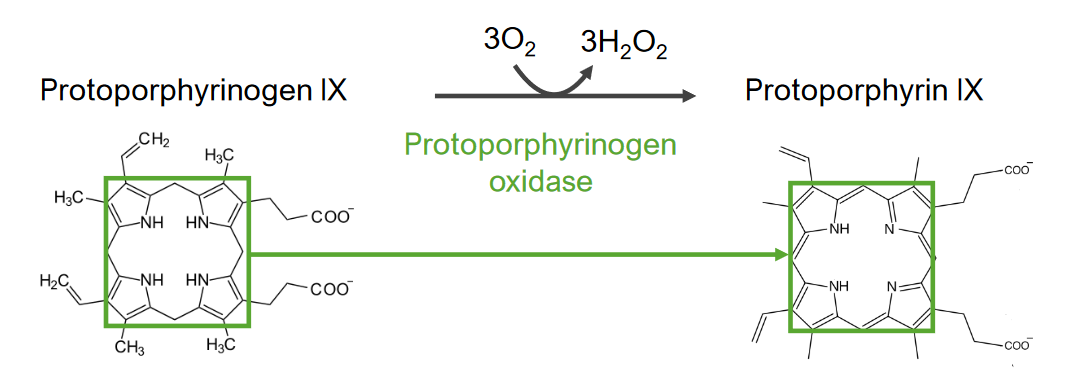
7.ª etapa do metabolismo do grupo heme:
Formação de protoporfirina a partir do protoporfirinogénio IX
A 8.ª etapa consiste na formação do grupo heme.
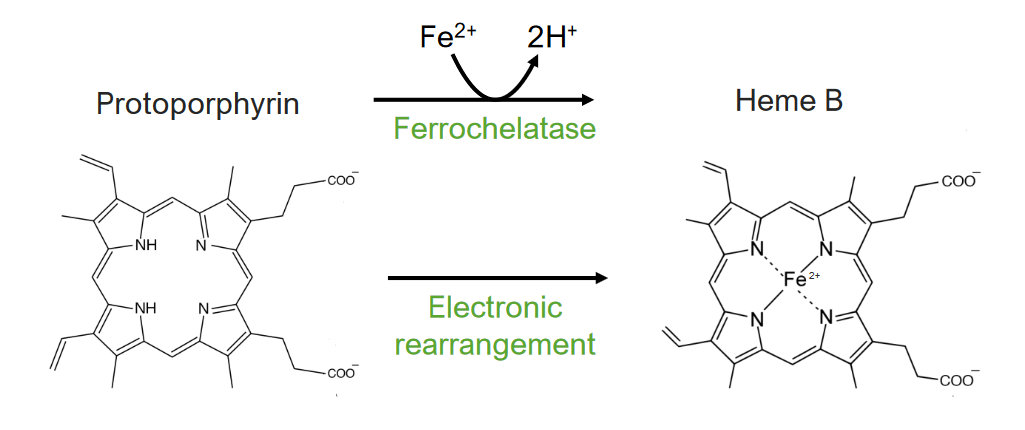
A 8.ª e última etapa do metabolismo do grupo heme:
Formação do grupo heme
| Etapa | Local do processo | Enzima | Doença associada com a presença de mutações genéticas enzimáticas |
|---|---|---|---|
| 1. Síntese de ácido aminolevulínico | Mitocôndria | Sintase do ácido aminolevulínico |
|
| 2. Formação de porfobilinogénio ( PBG PBG Heme Metabolism) | Citosol | Desidratase do ácido aminolevulínico ou sintase do PBG PBG Heme Metabolism | Porfiria da desidratase do ácido aminolevulínico |
| 3. Formação de hidroximetilbilano ( HMB HMB Excessive menstrual blood loss (objectively defined as > 80 mL blood loss/cycle). Can be based on heavy flow, as determined by the patient Abnormal Uterine Bleeding) | Desaminase do PBG PBG Heme Metabolism/Sintase do HMB HMB Excessive menstrual blood loss (objectively defined as > 80 mL blood loss/cycle). Can be based on heavy flow, as determined by the patient Abnormal Uterine Bleeding | Porfiria aguda intermitente | |
| 4. Formação de uroporfirinogénio (UPG) | Sintase do UPG III | Porfiria eritropoiética congénita | |
| 5. Síntese de coproporfirinogênio (CPG) III | Desxarboxilase do UPG | Porfiria cutânea tardia e porfiria hepatoeritropoiética | |
| 6. Síntese de protoporfirinogénio (PPG) | Mitocôndria | Oxidase Oxidase Neisseria do CPG | Coproporfiria hereditária |
| 7. Formação de protoporfirina (PP) | Oxidase Oxidase Neisseria do protoporfirinogénio | Porfiria variegata | |
| 8. Formação do grupo heme | Ferroquelatase/sintase do heme | Protoporfiria eritropoiética |
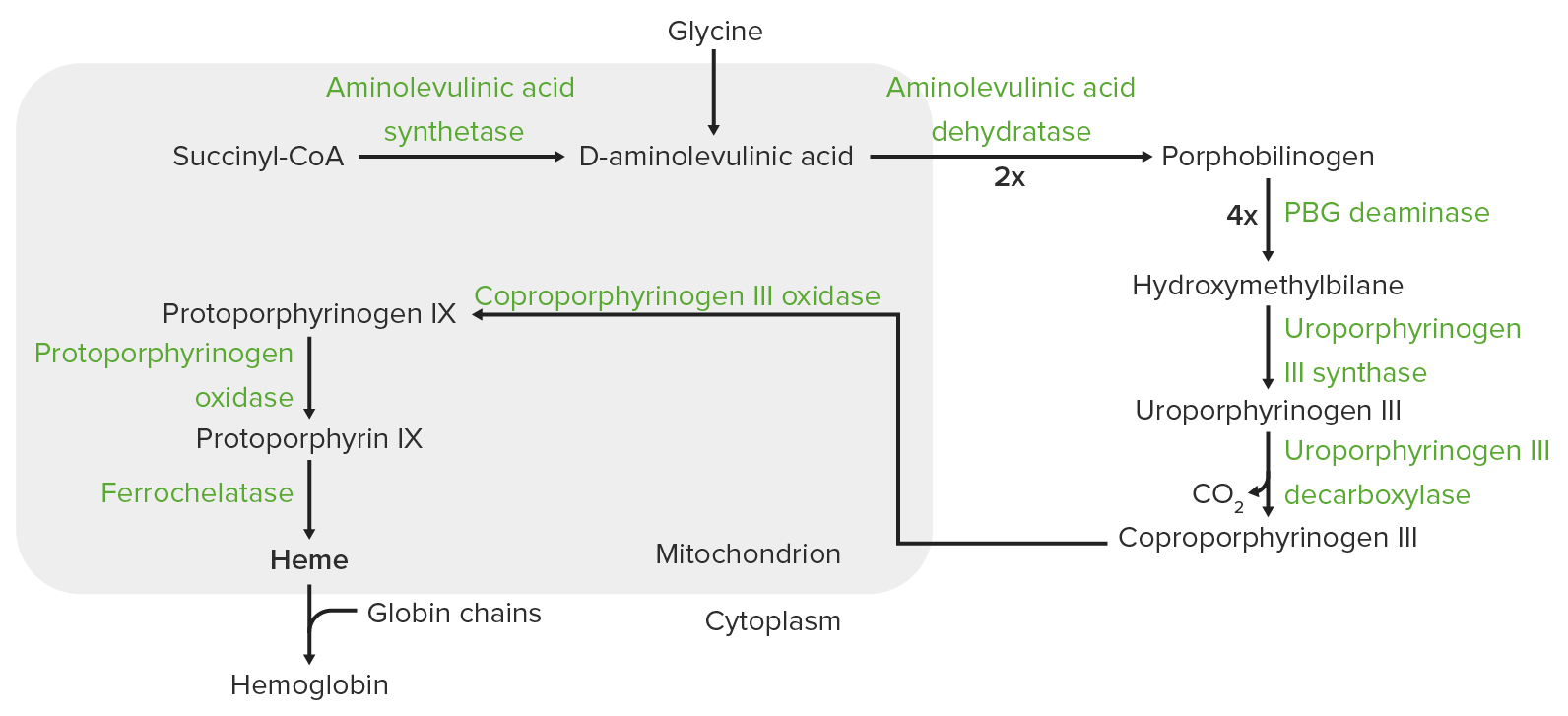
Síntese do grupo Heme:
O processo de síntese do grupo heme ocorre na mitocôndria e no citoplasma.
Nas mitocôndrias, a succinil coenzima A (CoA) combina-se com a glicina para formar o ácido aminolevulínico.
Esta reação é catalisada pela sintase do ácido aminolevulínico. O ácido aminolevulínico sai para o citoplasma, onde 2 moléculas de ácido aminolevulínico condensam-se para produzir o porfobilinogénio (PBG). As etapas subsequentes levam à formação do coproporfirinogénio III, que é transportado de volta para a mitocôndria. A oxidase facilita a conversão do coproporfirinogénio III em protoporfirinogénio IX, que é convertido em protoporfirina IX. O ferro é inserido na protoporfirina IX, formando o grupo heme (catalisado pela ferroquelatase).
O grupo heme decompõem-se, com consequente formação de produtos finais como os pigmentos biliares, como a bilirrubina excretada pela bílis. As etapas de catabolismo do grupo heme são as seguintes:
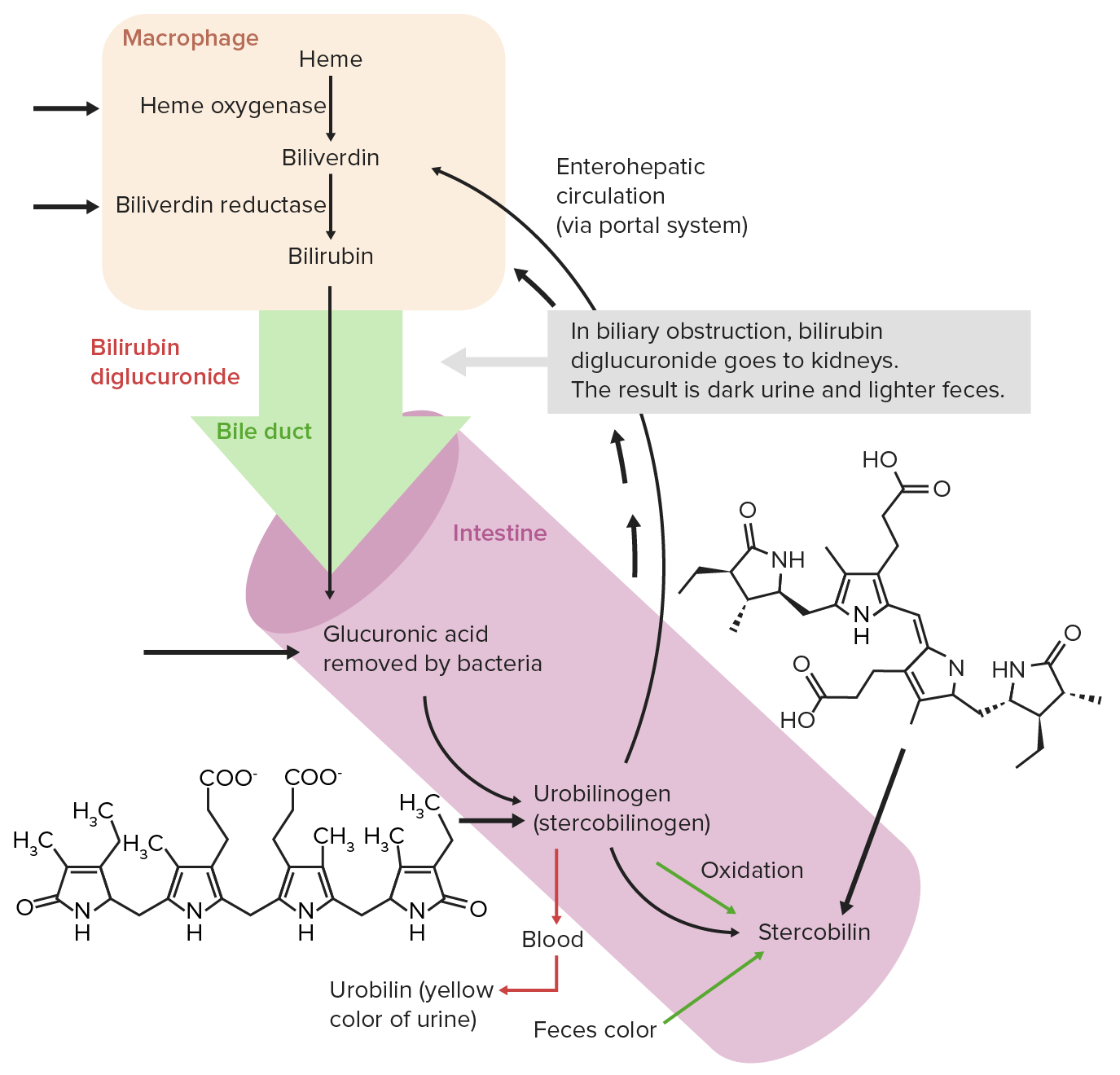
Circulação extra-hepática normal da bilirrubina
Imagem por Lecturio. Licença: CC BY-NC-SA 4.0Absorção de ferro:
Transporte do ferro:
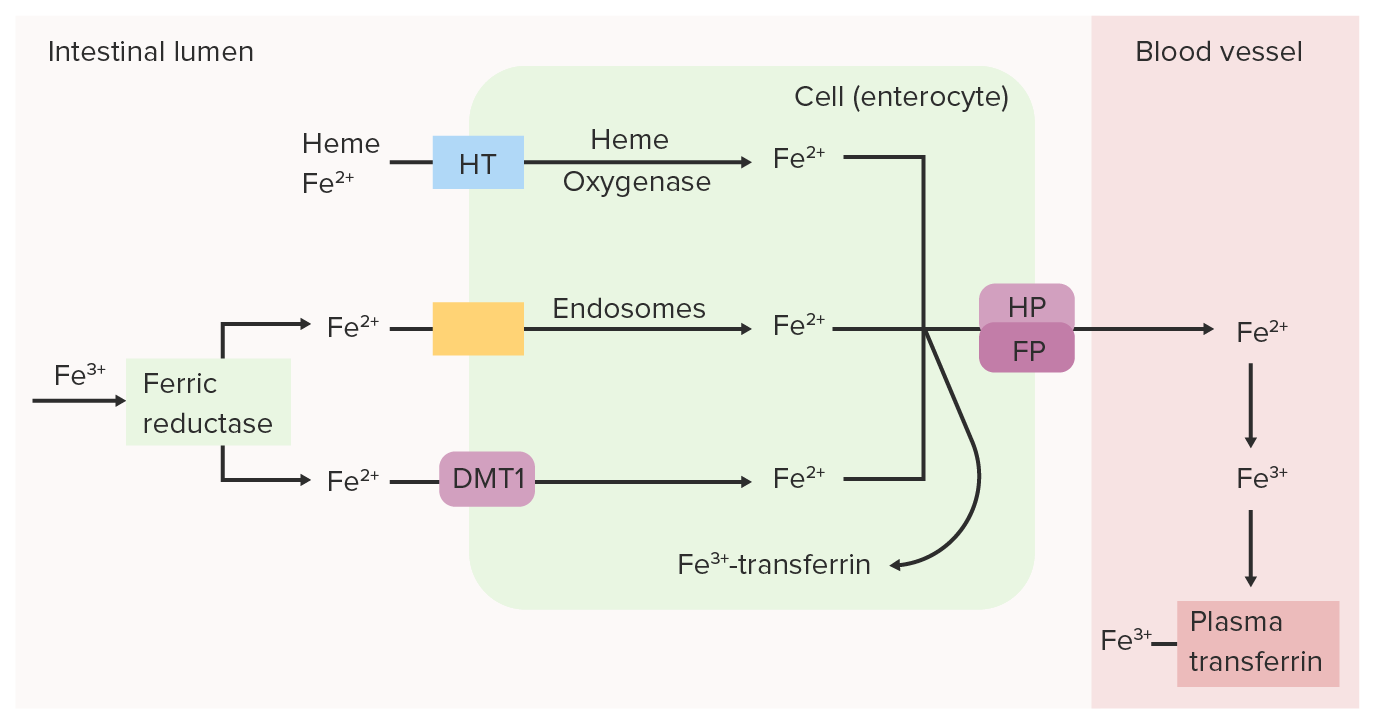
A redutase férrica (Fe 3+) intestinal reduz o Fe 3+ (férrico) a Fe 2+ (ferroso). O Fe 2+ é transportado do lúmen para a célula epitelial intestinal através do transportador de metal divalente 1 (DMT1, pela sigla em inglês), transportador do heme (HT) e/ou endossomas. O Fe 2+ pode ser convertido novamente em Fe 3+ e ligado à transferrina dentro da célula intestinal ou pode ser transportado para o sangue pela ferroportina (FP) e hefestina (HP). O ferro oxidado (Fe 3+), que se liga à transferrina plasmática, é transportado pela circulação até os tecidos.
Imagem por Lecturio.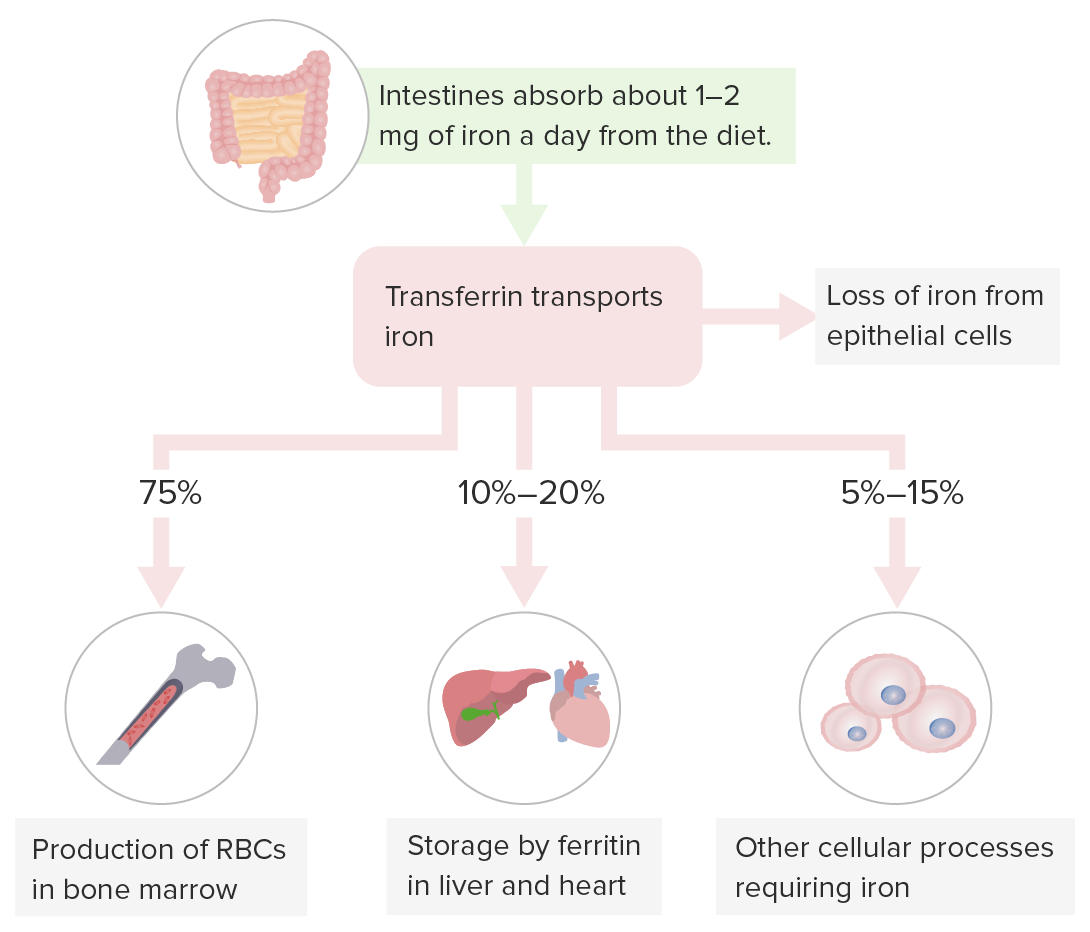
Armazenamento do ferro:
A transferrina transporta o ferro para a hematopoiese na medula óssea, armazenamento no fígado (local de armazenamento primário), e noutros órgãos, e para processos celulares que requerem ferro.
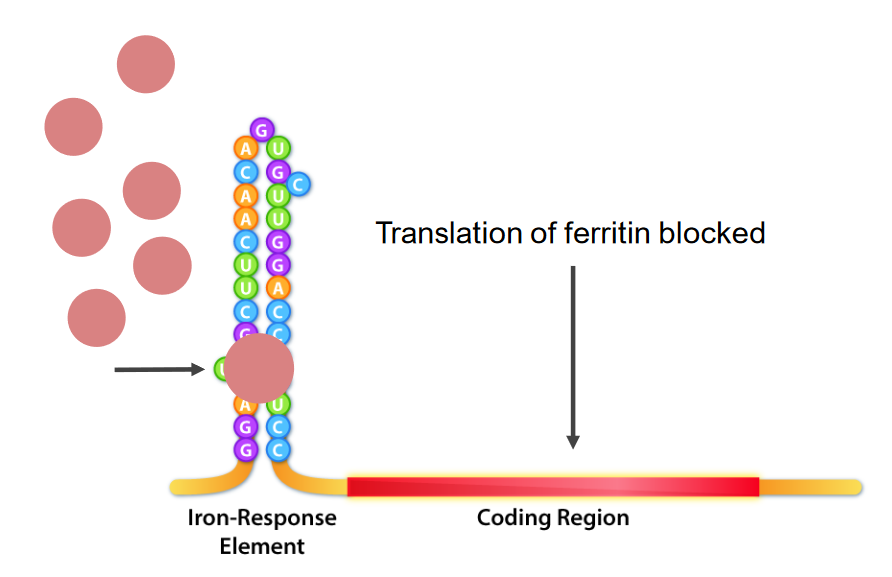
Interação das proteínas de ligação do elemento de resposta ao ferro (IRE, pela sigla em inglês) (IRBPs, pela sigla em inglês; estruturas redondas com cor rosa) e IRE, que é a estrutura da ansa da haste na extremidade 5′ do mRNA. No défice de ferro, os IRBPs ligam-se aos IREs, causando a inibição da tradução do mRNA da ferritina.
Imagem por Kevin Ahern.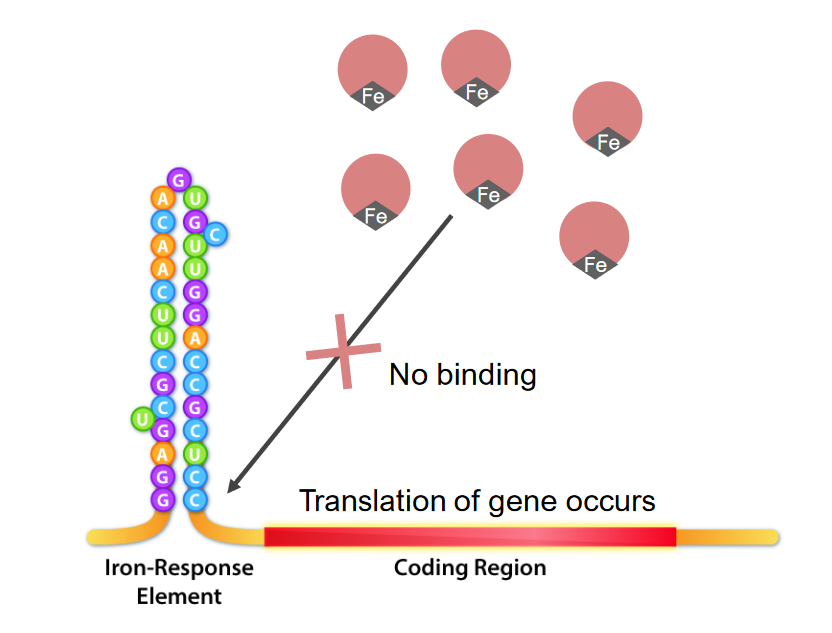
Interação de elementos de resposta do ferro e proteínas de ligação aos IRE (IRBPs) no caso de excesso de ferro:
O ferro liga-se aos IRBPs, tornando-os indisponíveis para se ligarem à ferritina. Isso permite a manutenção da tradução.
Em certas condições é necessário ocorrer uma diminuição ou aumento na absorção de ferro e nos níveis de ferro circulante. Esta via é regulada pela hepcidina:
