El hemo es una porfirina que contiene hierro (que está formada por 4 grupos pirrol), sintetizada principalmente en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la médula ósea y el hígado. El hemo es un componente de muchas sustancias cruciales, incluyendo los LOS Neisseria citocromos, mioglobina y hemoglobina. Las funciones biológicas incluyen el transporte de gases (e.g., O2) y la transferencia de electrones. La biosíntesis del hemo es un proceso de 8 pasos iniciado por la síntesis de ácido aminolevulínico. La disponibilidad de hierro afecta la producción del hemo, ya que el último paso implica la inserción de iones ferrosos. El hierro se obtiene de la dieta y de la descomposición de los LOS Neisseria productos que contienen hemo. En EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el proceso de catabolismo, el hemo se convierte en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum pigmentos biliares, de los LOS Neisseria cuales se excreta la bilirrubina. Las mutaciones que involucran a las enzimas en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la síntesis del hemo conducen a un grupo de trastornos conocidos como porfirias y un defecto en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el catabolismo del hemo causa hiperbilirrubinemias.
Last updated: Apr 25, 2025
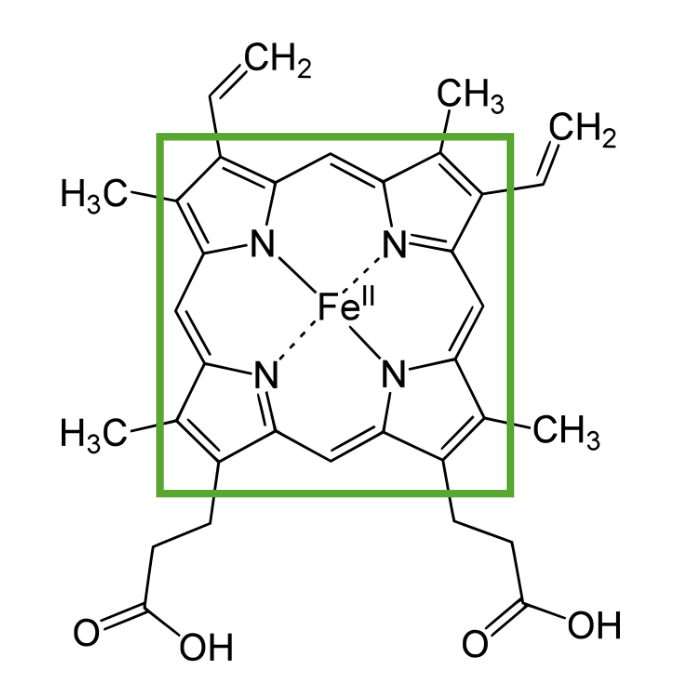
Estructura del hemo que muestra un anillo de porfirina con un elemento ferroso en el centro
Imagen por Lecturio.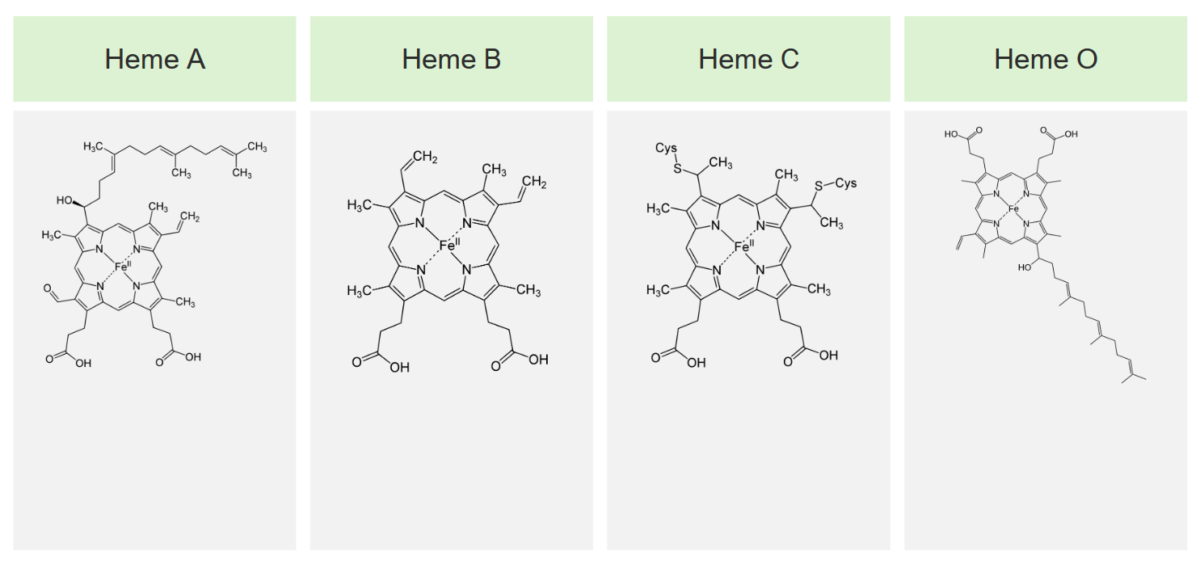
Los 4 tipos principales de hemo y sus estructuras
Imagen por Lecturio.El hemo se sintetiza en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria normoblastos, pero no en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria eritrocitos maduros. La biosíntesis del hemo se realiza en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum 8 pasos.
El paso 1 es la síntesis de ácido aminolevulínico.
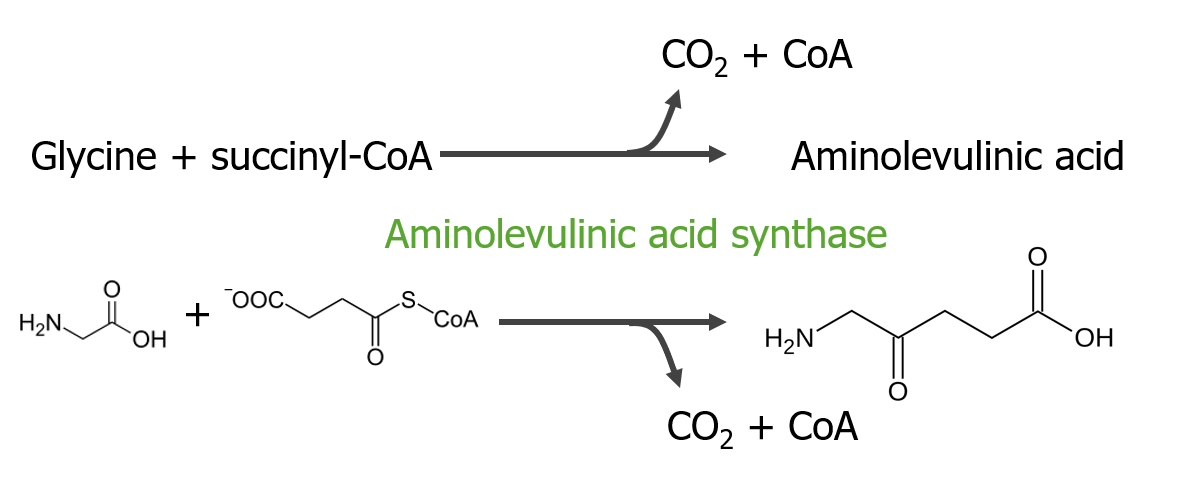
Paso 1 del metabolismo del hemo
Imagen por Lecturio.El paso 2 es la formación de porfobilinógeno.
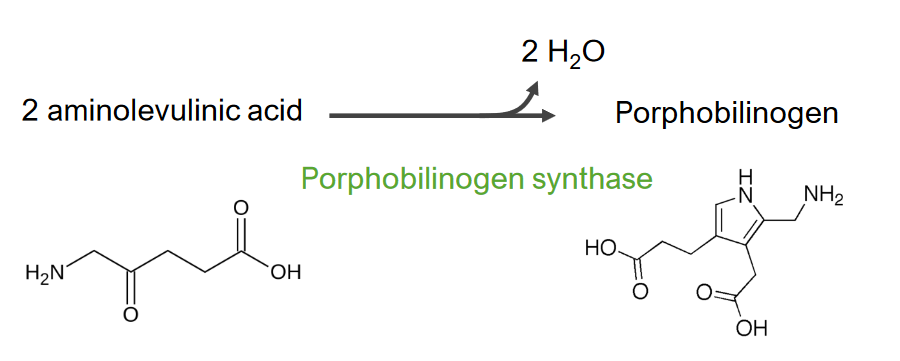
Paso 2 del metabolismo del hemo
Formación de porfobilinógeno
El paso 3 es la formación de hidroximetilbilano.
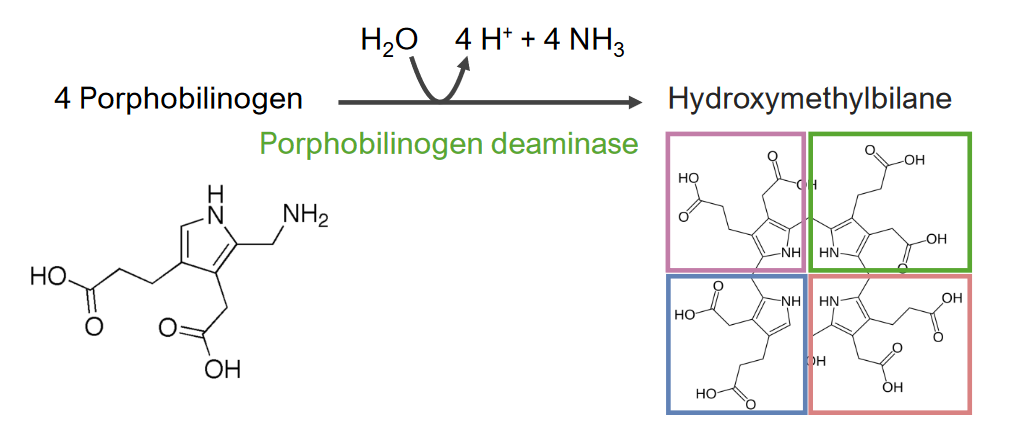
Paso 3 del metabolismo del hemo:
Formación de hidroximetilbilano
El paso 4 es la formación de uroporfirinógeno.
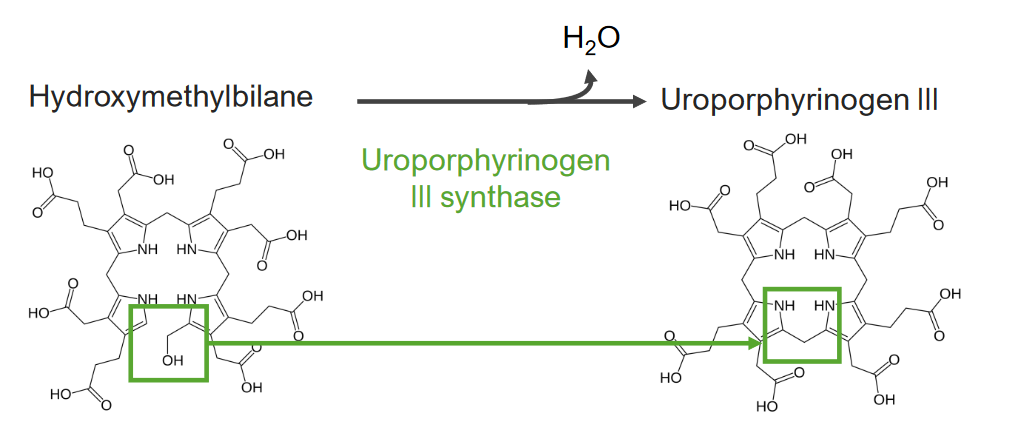
Paso 4 del metabolismo del hemo:
Formación de uroporfirinógeno
El paso 5 es la síntesis de coproporfirinógeno III.
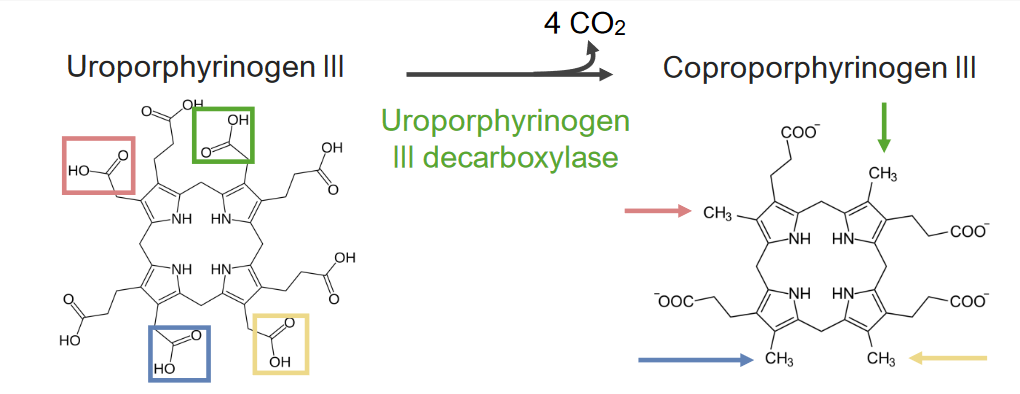
Paso 5 del metabolismo del hemo:
Formación de coproporfirinógeno III
El paso 6 es la síntesis de protoporfirinógeno.
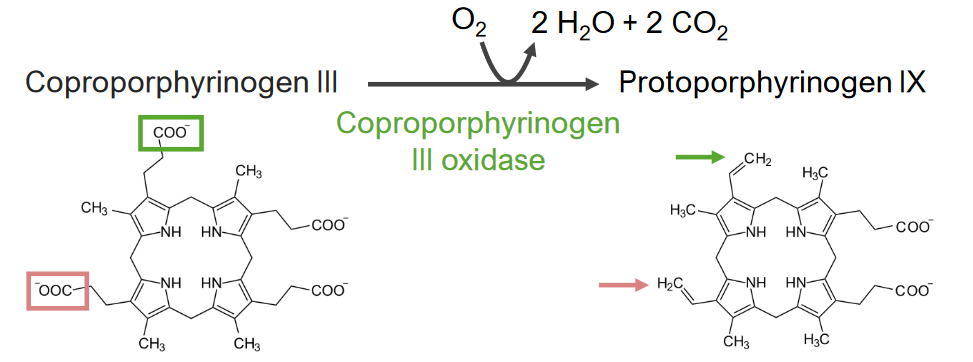
Paso 6 del metabolismo del hemo:
Síntesis de protoporfirinógeno
El paso 7 es la generación de protoporfirina.
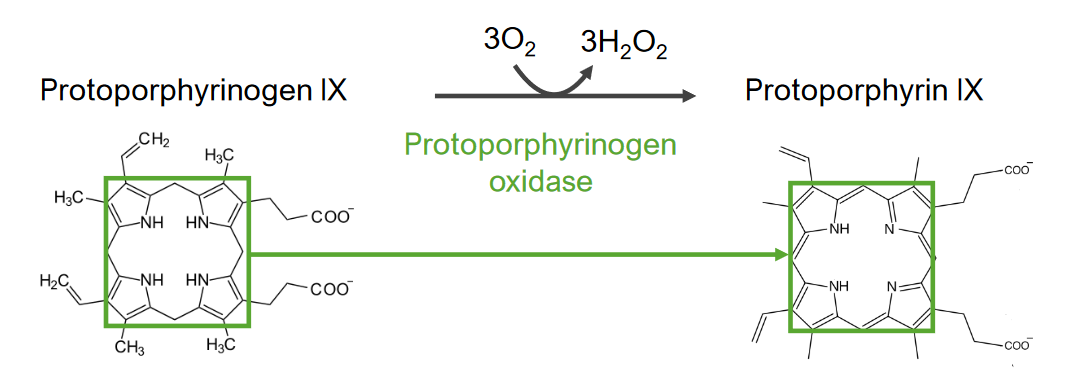
Paso 7 del metabolismo del hemo:
Generación de protoporfirina a partir del protoporfirinógeno IX
El paso 8 es la generación de hemo.
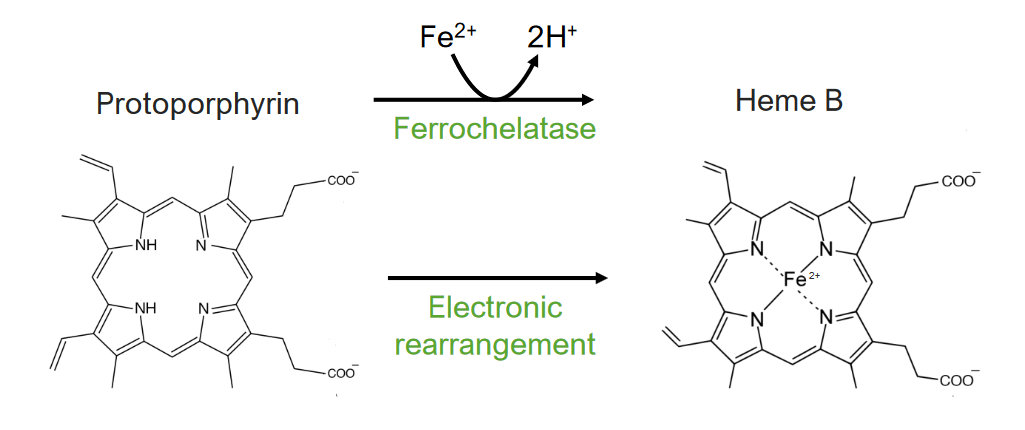
El 8vo y último paso del metabolismo del hemo:
Formación del hemo
| Paso | Sitio del proceso | Enzima | Enfermedad asociada con mutaciones del gen de la enzima |
|---|---|---|---|
| 1. Síntesis de ácido aminolevulínico (ALA) | Mitocondria | Ácido aminolevulínico sintasa |
|
| 2. Formación de porfobilinógeno ( PBG PBG Heme Metabolism) | Citosol | Ácido aminolevulínico deshidratasa o porfobilinógeno sintasa | Porfiria del ácido aminolevulínico deshidratasa |
| 3. Formación de hidroximetilbilano | Porfobilinógeno desaminasa/hidroximetilbilano sintasa | Porfiria aguda intermitente | |
| 4. Formación de uroporfirinógeno | Uroporfirinógeno III sintasa | Porfiria eritropoyética congénita | |
| 5. Síntesis de coproporfirinógeno III | Uroporfirinógeno descarboxilasa | Porfiria cutánea tardía y porfiria hepatoeritropoyética | |
| 6. Síntesis de protoporfirinógeno | Mitocondria | Coproporfirinógeno oxidasa | Coproporfiria hereditaria |
| 7. Generación de protoporfirina | Protoporfirinógeno oxidasa | Porfiria variegata | |
| 8. Generación de hemo | Ferroquelatasa/hem sintasa | Protoporfiria eritropoyética |
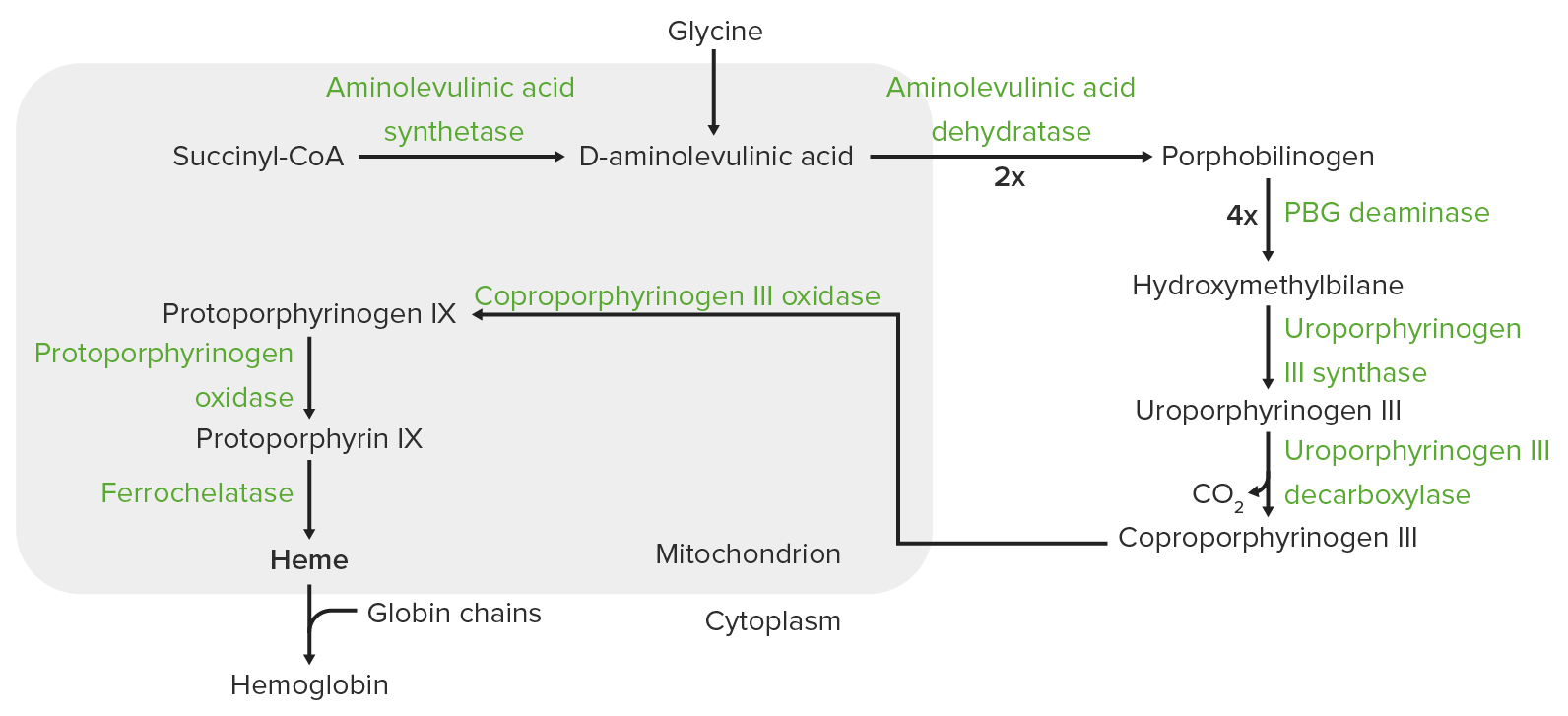
Síntesis del hemo:
El proceso de síntesis del hemo tiene lugar en la mitocondria y el citosol.
En las mitocondrias, la succinil coenzima A (CoA) se combina con la glicina para formar ácido aminolevulínico.
Esta reacción es catalizada por la enzima ácido aminolevulínico sintasa. El ácido aminolevulínico sale al citosol, donde 2 moléculas de ácido aminolevulínico se condensan para producir porfobilinógeno (PBG). Los pasos posteriores conducen a la formación de coproporfirinógeno III, que se transporta de regreso a la mitocondria. La oxidasa facilita la conversión de coproporfirinógeno III en protoporfirinógeno IX, que luego se convierte en protoporfirina IX. El hierro ferroso se inserta en la protoporfirina IX, formando hemo (catalizado por la enzima ferroquelatasa).
El hemo se descompone, dando como resultado pigmentos biliares como productos finales y la bilirrubina se excreta a través de la bilis. Los LOS Neisseria pasos del catabolismo del hemo son:
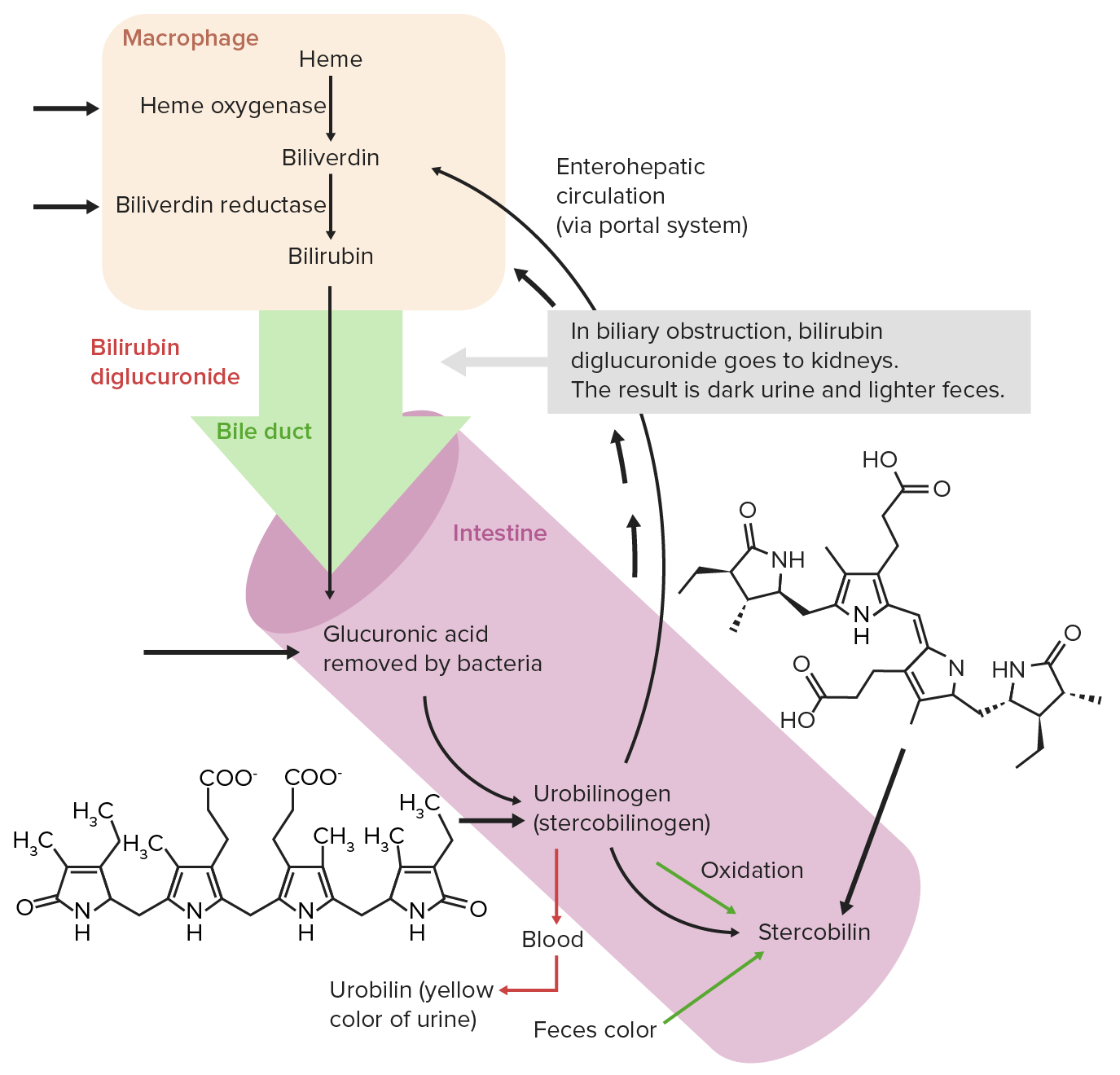
Circulación extrahepática normal de la bilirrubina
Imagen por Lecturio. Licencia: CC BY-NC-SA 4.0Absorción del hierro:
Transporte del hierro:
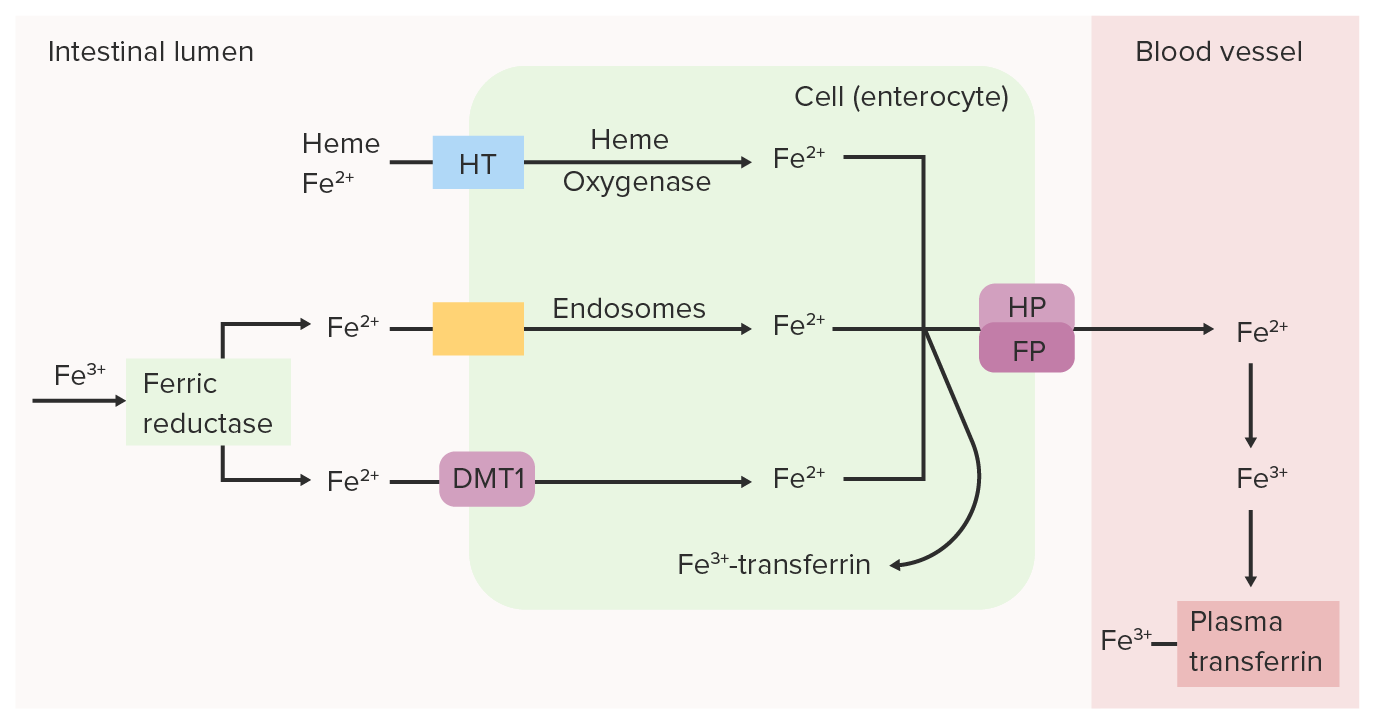
La reductasa férrica (Fe3+) intestinal reduce el Fe3+ (férrico) a Fe2+ (ferroso). El Fe2+ se transporta desde la luz hacia la célula epitelial intestinal a través del transportador de metal divalente 1 (DMT1), el transportador de hemo (HT) y/o los endosomas. El Fe2+ puede volver a convertirse en Fe3+ y unirse a la transferrina dentro de la célula intestinal o puede transportarse a la sangre mediante ferroportina (FP) y hefestina (HP). El hierro oxidado (Fe3+), que se une a la transferrina plasmática, se transporta a través de la circulación hasta los tejidos.
Imagen por Lecturio.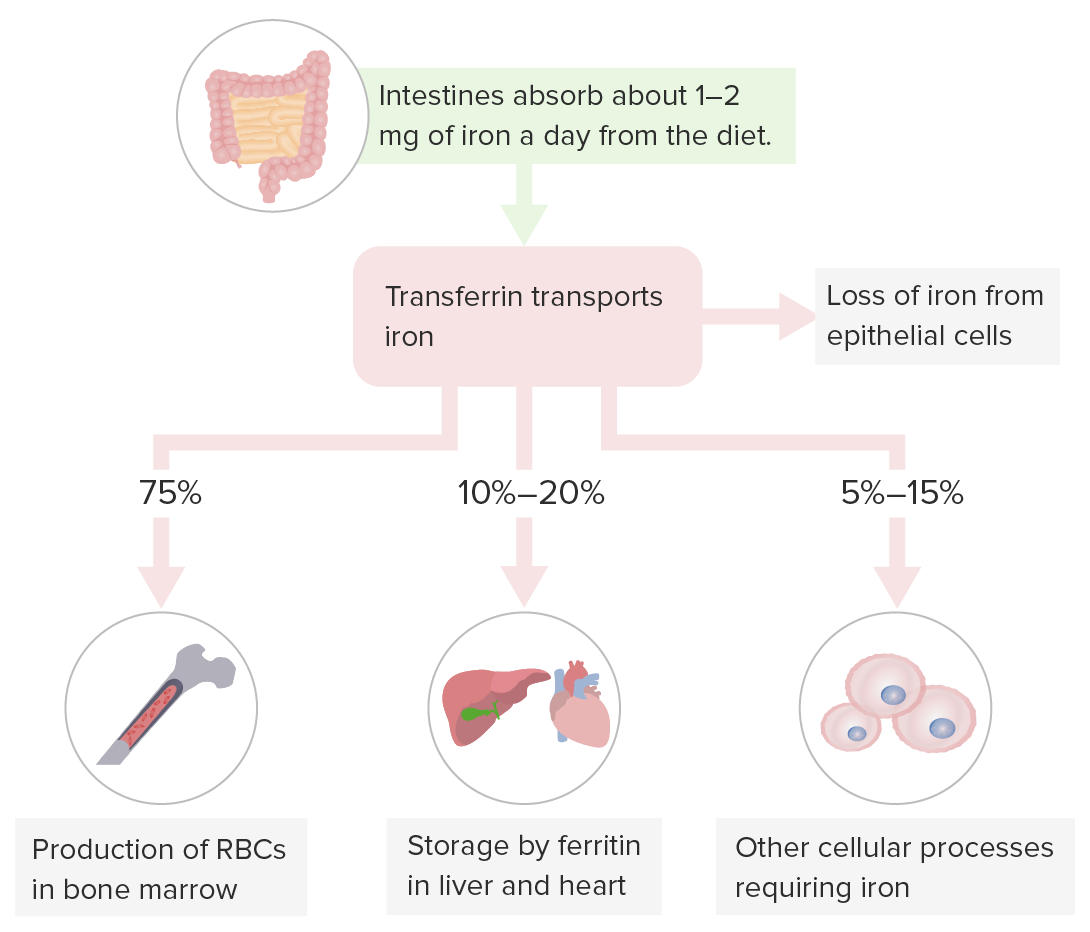
Almacenamiento del hierro:
La transferrina transporta hierro para la hematopoyesis en la médula ósea, el almacenamiento de hierro en el hígado (sitio de almacenamiento principal) y otros órganos y los procesos celulares que requieren hierro.
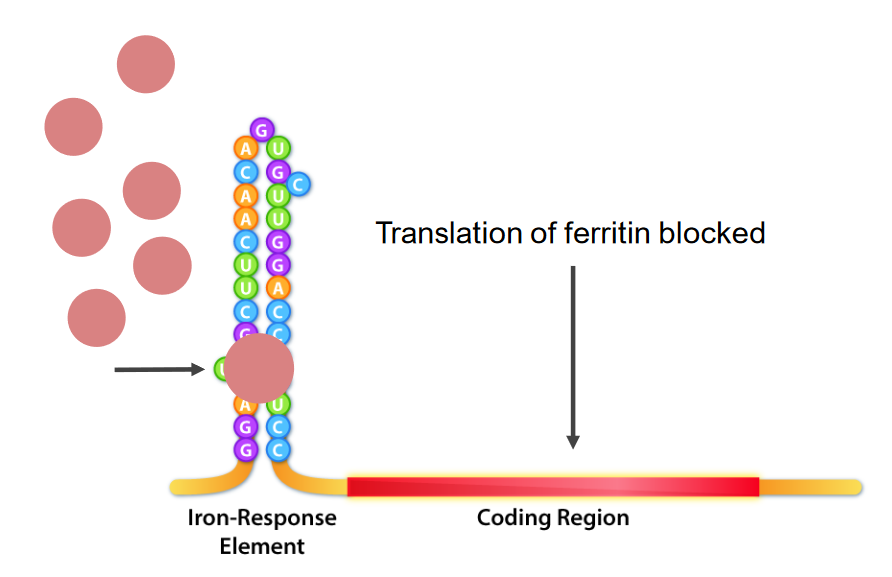
Interacción de las proteínas de unión a los elementos de respuesta al hierro (IRE) (IRBP; estructuras redondas rosadas) e IRE, que son las estructuras de bucle de tallo en el extremo 5 ‘del ARNm. En la deficiencia de hierro, las IRBP se unen a los IRE, lo que provoca la inhibición de la traducción del ARNm de ferritina.
Imagen por Kevin Ahern.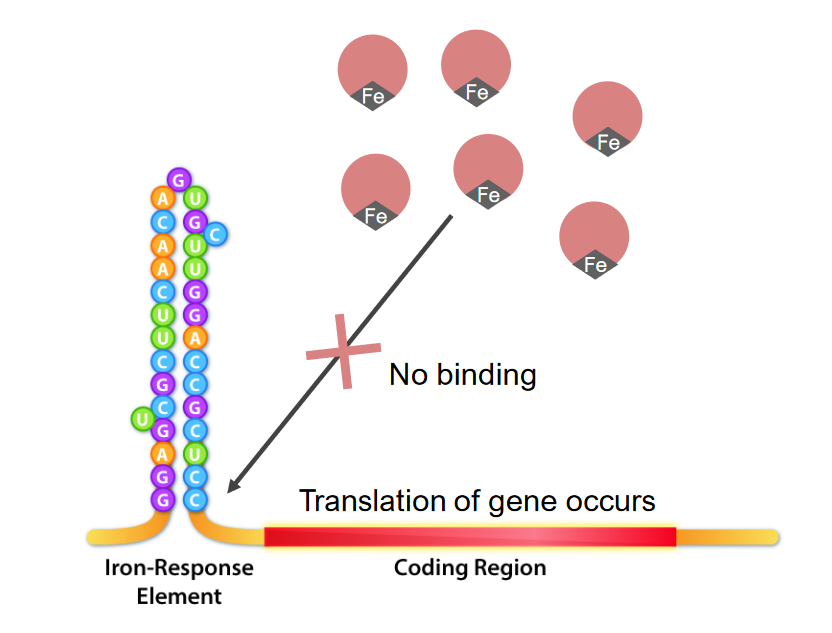
Interacción de los elementos de respuesta al hierro y las proteínas de unión a IRE (IRBP) en exceso de hierro:
El hierro se une a las IRBP, lo que las hace incapaces de unirse a la ferritina IRE. Esto permite que la traducción continúe.
Ciertas condiciones requieren una disminución o aumento en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la absorción de hierro y el hierro circulante, una vía regulada por la hepcidina: