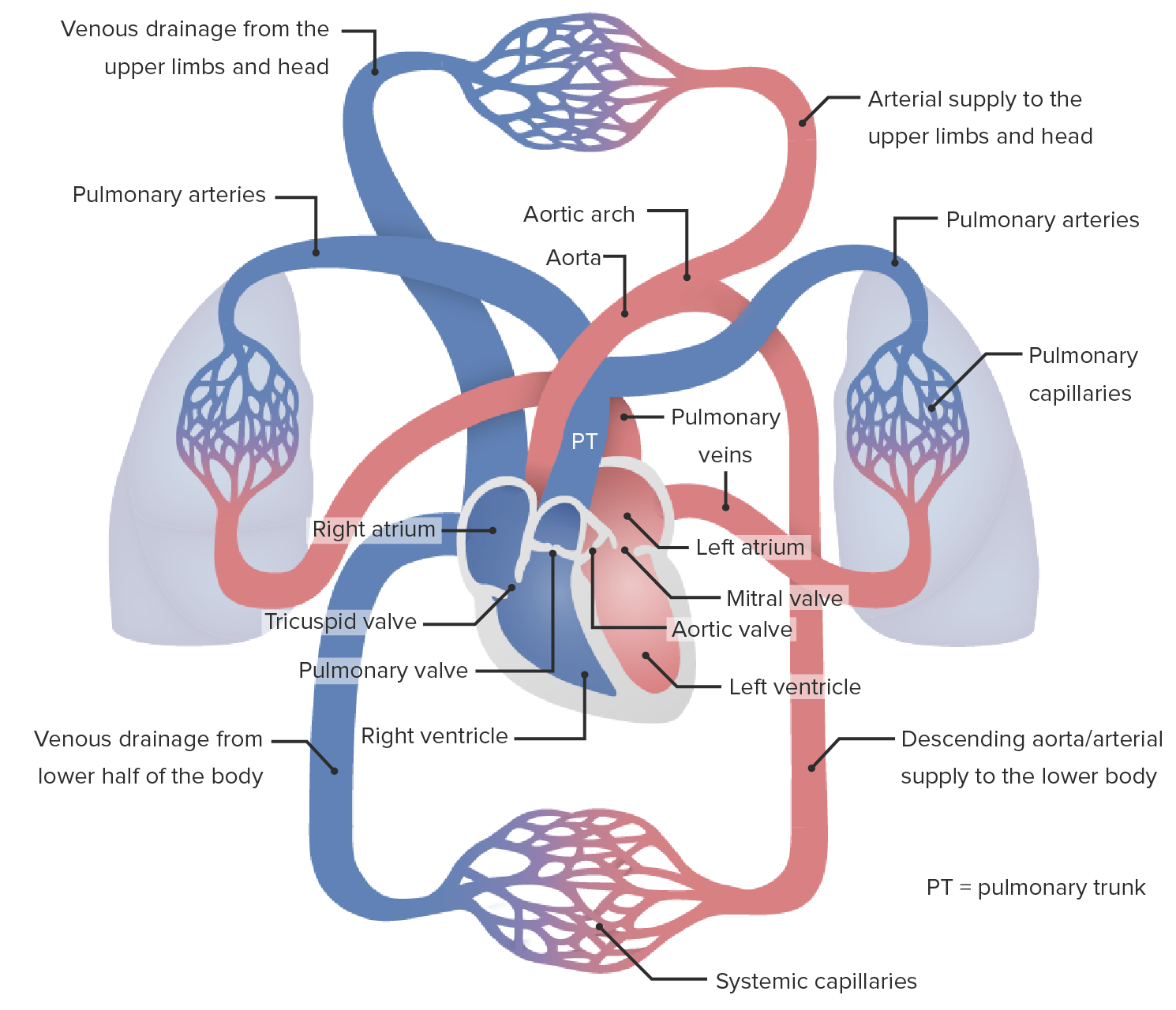
A hipertensão pulmonar (HP) ou hipertensão arterial pulmonar (HAP) é caracterizada por elevação da pressão arterial pulmonar, que pode levar a insuficiência cardíaca direita progressiva crónica. A hipertensão pulmonar é agrupada em 5 categorias baseadas na etiologia, que incluem HAP primária e HP devido a doença cardíaca, doença pulmonar ou hipóxica, doença tromboembólica crónica e etiologia multifatorial ou pouco claras. Os pacientes normalmente apresentam dispneia inicialmente durante o exercício e depois em repouso. Para fazer o diagnóstico pode ser necessário recorrer ao ecocardiograma, ECGECGAn electrocardiogram (ECG) is a graphic representation of the electrical activity of the heart plotted against time. Adhesive electrodes are affixed to the skin surface allowing measurement of cardiac impulses from many angles. The ECG provides 3-dimensional information about the conduction system of the heart, the myocardium, and other cardiac structures. Electrocardiogram (ECG), radiografia de tórax, testesTestesGonadal Hormones de função pulmonar, cintigrafia de ventilação-perfusão, testesTestesGonadal Hormones laboratoriais para condições associadas à HAP, e/ou cateterismo cardíaco. O tratamento é muitas vezes complexo e tem como objetivo tratar a etiologia subjacente. Várias classes de vasodilatadores podem ser usadas em pacientes com HAP primária, incluindo bloqueadores dos canais de cálcio e prostaglandinas vasoativas.
A hipertensão pulmonar (HP) define-se como pressão arterial pulmonar elevada.
Pressão arterial pulmonar elevada consistente com a HP:
Definição histórica:
≥ 25 mm Hg em repouso
≥ 30 mm Hg durante o exercício
Consenso do 6.º World Symposia on Pulmonary Hipertension em 2018: pressão arterial pulmonar média > 20 mm Hg em repouso
Pressão arterial pulmonar normal: 10-14 mm Hg
Pode ser primária (pouco comum) ou secundária (comum):
A HP primária é referida como hipertensão arterial pulmonar (HAP).
A HP secundária pode ser devido a doenças cardíacas crónicas, pulmonares ou sistémicas.
Epidemiologia
A HP secundária é muito maisMAISAndrogen Insensitivity Syndrome comum do que a primária. A epidemiologia da HP secundária é semelhante à condição subjacente.
HP/HAP primária:
Prevalência: aproximadamente 15 casos em 1 milhão de adultos
A OMS classificou a HP em 5 categorias com base na etiologia:
Grupo 1: HAP
O grupo 1 refere-se a casos de aumento da pressão arterial pulmonar na ausência de doença cardíaca ou pulmonar subjacente e era anteriormente conhecido como (e ainda pode ser conceptualizado como) HPprimária. As etiologias do grupo 1 incluem:
HAP familiar: devido a mutações no geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics do recetor da proteína morfogénica óssea – 2(BMPR2), várias outras mutações raras, ou causas desconhecidas
Fármacos ou toxinas:
Supressores de apetite:
Anfetaminas
Fenfluramina
Aminorex
Óleo de colza
Cocaína
Associada a:
Doenças do tecido conjuntivo (principalmente esclerodermia)
Doenças cardíacas do lado esquerdo podem causar aumento da pressão a montante na vasculatura pulmonar, levando à HP. É a causa maisMAISAndrogen Insensitivity Syndrome comum de HP no geral. As atiologias incluem:
Cardiopatia auricular ou ventricular esquerda
Doença cardíaca valvular esquerda
Obstrução das vias de entrada ou saída do coração esquerdo (congénita ou adquirida)
Cardiomiopatias congénitas
Grupo 3: HP por doença pulmonar crónica e/ou hipoxia
A hipoxia leva à vasoconstrição fisiológica da vasculatura pulmonar para evitar o desequilíbrio ventilação-perfusão. Como resultado, a hipoxia crónica, bem como as doenças pulmonares destrutivas, podem levar à HP crónica. As etiologias incluem:
Doenças pulmonares de padrão misto (restritivos e obstrutivos)
Distúrbios respiratórios do sono (por exemplo, apneia obstrutiva do sono)
Distúrbios da hipoventilação alveolar:
Síndrome de hipoventilação-obesidade
Síndrome de hipoventilação alveolar central congénita
Disfunção hipotalámica
Exposição crónica a altitudes elevadas
Grupo 4: HP por doença tromboembólica crónica
Os casos do grupo 4 são diagnosticados quando há um aumento da pressão arterial pulmonar com documentação de obstrução arterial pulmonar. As etiologias incluem:
Tromboembolia pulmonar crónica
Embolia pulmonar não-trombótica:
Embolias neoplásicas (manifestações tardias de certas neoplasias malignas, com embolização das próprias partículas tumorais)
Grupo 5: HP devido a causas pouco claras ou multifatoriais
A hipertensão pulmonar é classificada em HP do Grupo 5 quando a elevação da pressão arterial pulmonar está associada a uma doença sistémica, em que a relação causal não está claramente entendida ou pensa-se ser multifatorial. As etiologias incluem:
Distúrbios hematológicos:
AnemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica crónica:
AnemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types falciforme
Em última análise, leva a cor pulmonaleCor PulmonaleCor pulmonale is right ventricular (RV) dysfunction caused by lung disease that results in pulmonary artery hypertension. The most common cause of cor pulmonale is chronic obstructive pulmonary disease. Dyspnea is the usual presenting symptom. Cor Pulmonale(insuficiência cardíaca direita).
Pressão arterial pulmonar média = (Q x RVP) + pressão em cunha pulmonar
Q: débito cardíaco direito
RVP: resistência vascular pulmonar
A pressão em cunha pulmonar estima a pressão auricular esquerda.
O ↑ da pressão arterial pulmonar pode ser devida a:
O aumento da resistência vascular pulmonar é a causa principal da HP na maioria dos casos e pode ser devido a:
Vasculopatias oclusivas das pequenas artérias/arteriolas pulmonares: remodelam a vasculatura e alteram o tom (por exemplo, HAP idiopática)
↓ Na área do leito vascular pulmonar:
Embolia pulmonar
Doença pulmonar intersticial
Vasoconstrição hipóxica:
Síndromes de hipoventilação
Doença pulmonar parenquimatosa
↑ Pressão venosa pulmonar:
Doença da válvula mitral
Disfunção ventricular esquerda
Pericardite constritiva
Cardiomiopatia restritiva
Obstrução venosa pulmonar
Aumento do fluxo através da vasculatura pulmonar
Tipicamente, o aumento do fluxo desencadeia vasodilatação da vasculatura pulmonar. Nos casos em que este aumento de fluxo é crónico, a HP pode desenvolver-se. Aumentos crónicos do fluxo também podem provocar alterações vasculares e levar ao aumento da resistência vascular pulmonar.
Defeitos cardíacos congénitos com shunt da esquerda para a direita:
Defeitos do septo interauricular
Defeitos do septo interventricular
Ducto arterial patente
Cirrose hepática
AnemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types crónica
Malformações arteriovenosas
Mutações genéticas
A HAP familiar é maisMAISAndrogen Insensitivity Syndrome frequentemente devida a mutações no geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of GeneticsBMPR2.
BMPR2
80% dos casos familiares são devidos a uma mutação inativadora no BMPR2.
BMPR2 normalmente inibe a proliferação do músculo liso vascular.
Com o BMPR2 inativado, os pacientes são incapazes de prevenir a proliferação do músculo liso vascular → HAP
Foram identificadas várias outras mutações raras.
Patogénese por grupo
Tabela: Patogénese de HP por grupo
Grupo 1: “primária”.
Vasoconstrição do sistema arterial pulmonar
Proliferação das células vasculares
Fibrose
Grupo 2: devido a doença cardíaca esquerda
↑ pressões das câmaras cardíacas esquerdas (↑ pressão em cunha pulmonar) → ↑ pressão arterial pulmonar média
Remodelação vascular pulmonar
Redução da complacência da vasculatura pulmonar
Grupo 3: devido a doença pulmonar ou hipóxica
A hipoxia desencadeia a vasoconstrição pulmonar para prevenir o desequilíbrio ventilação-perfusão através dos seguintes mecanismos:
↓ produção NO (um vasodilatador)
Disfunção dos canais de potássio dependentes de voltagem → contração do músculo liso pulmonar
↑ Atividade da fosfolipase A2 → aumento das substâncias vasoconstritoras: prostaglandinas vasoconstritoras, tromboxanos, leucotrienos
↑ Endotelina (um vasoconstritor)
Destruição vascular devido a fibrose parenquimatosa progressiva
Inflamação vascular
Grupo 4: devido a tromboembolia crónica
Semelhante ao grupo 3
Apresentação Clínica
História clínica
Sintomas:
Dispneia com o esforço que progride para dispneia em repouso (sintoma inicial)
Fadiga
Dor torácica (angina)
Síncope com o exercício
Considerações dos antecedentes médicos:
DPOC/enfisema
Doença pulmonar intersticial
Cardiopatia
AnemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types falciforme
Viagens para regiões endémicas de esquistossomose
Estados de hipercoagulação/história de doença tromboembólica
Exame físico
Os resultados dos exames consistentes com HP incluem:
S4S4Heart Sounds: ↑ da resistência ao enchimento ventricular devido à ↓ da complacência ventricular
Sopros:
Insuficiência tricúspide: sopro holossistólico ouvido no bordo esternal esquerdo
Estenose mitral: sopro diastólico ouvido no ápex
Estenose aórtica: murmúrio médio-sistólico ouvido melhor no 2º espaço intercostal direito, irradiando para a carótida
Sinais de insuficiência cardíaca do lado direito:
EdemaEdemaEdema is a condition in which excess serous fluid accumulates in the body cavity or interstitial space of connective tissues. Edema is a symptom observed in several medical conditions. It can be categorized into 2 types, namely, peripheral (in the extremities) and internal (in an organ or body cavity). Edema periférico
Ascite
Derrame pleural
Hepatomegalia
Diagnóstico
O diagnóstico de HAP do grupo 1 é tipicamente um diagnóstico de exclusão, após excluir etiologias dos grupos 2-5, através dos seguintes testesTestesGonadal Hormones:
Ecocardiograma
É o melhor teste inicial
Permite a avaliação de:
Tamanho e função ventricular e auricular
Função das válvulas (por exemplo, presença de estenose ou regurgitação)
Evidência de sobrecarga de volume em ambos os lados
Os achados podem incluir:
↑ Espessura das paredes ventriculares
Hipocinese ventricular
Regurgitação valvular
ECGECGAn electrocardiogram (ECG) is a graphic representation of the electrical activity of the heart plotted against time. Adhesive electrodes are affixed to the skin surface allowing measurement of cardiac impulses from many angles. The ECG provides 3-dimensional information about the conduction system of the heart, the myocardium, and other cardiac structures. Electrocardiogram (ECG):
Importante para:
Exclusão de HP devida a doença cardíaca esquerda
Avaliação da função cardíaca direita
Os achados de doença cardíaca direita podem incluir:
Taquicardia
Desvio direito do eixo
Ondas R verticais em V1-V3
Achados na radiografia do tórax:
Alargamento das artérias pulmonares centrais e dos ramos principais
Afunilamento dos vasos distais
Achados consistentes com doença cardíaca: cardiomegalia, edemaEdemaEdema is a condition in which excess serous fluid accumulates in the body cavity or interstitial space of connective tissues. Edema is a symptom observed in several medical conditions. It can be categorized into 2 types, namely, peripheral (in the extremities) and internal (in an organ or body cavity). Edema pulmonar
Achados consistentes com doença pulmonar: DPOC, doença pulmonar intersticial
Nota: A radiografia do tórax é frequentemente normal ou com achados mínimos.
Cintigrafia de ventilação-perfusão:
Ajuda a diferenciar o grupo 3 (doença pulmonar/hipóxica) do grupo 4 (doença tromboembólica crónica)
Os achados podem incluir:
Grupo 3: perfusão difusa com manchas
Grupo 4: defeitos segmentares de incompatibilidade ventilação-perfusão
Provas de função pulmonar:
Útil no diagnóstico de distúrbios pulmonares e/ou hipóxicos
Hemodinâmica das câmaras cardíacas e dos grandes vasos
Débito cardíaco
Shunts cardíacos
Outras causas de dispneia ou angina
Permite testar a vasorreatividade em pacientes com HAP do grupo 1:
Administrar substância vasodilatadora de curta duração (por exemplo, NO inalado) e avaliar a resposta.
Importante para o tratamento de pacientes com HAP do grupo 1
TestesTestesGonadal Hormones laboratoriais: podem ajudar a identificar outras causas de HP
Hemograma: para exclusão de anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types
TestesTestesGonadal Hormones de função hepática: para exclusão de doença hepática como causa dos sintomas
Rastreio de VIH
ANA: como rastreio de esclerodermia
Estudos laboratoriais para esquistossomose
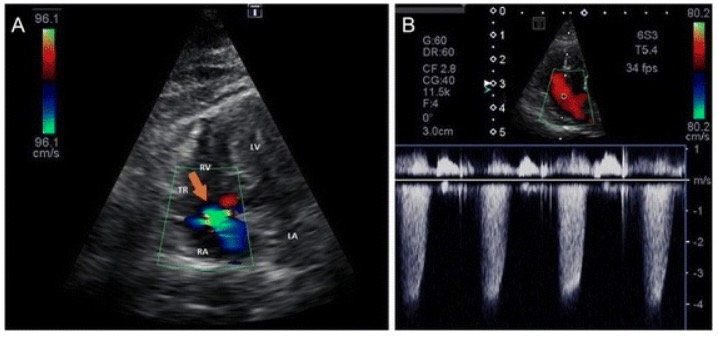
Insuficiência tricúspide: Regurgitação tricúspide de aproximadamente 4,2 m/seg, indicando um um pico do gradiente de pressão de regurgitação de aproximadamente 70 mm Hg (hipertensão pulmonar moderada) A: Imagem Doppler de regurgitação tricúspide B: Imagem de Doppler contínuo a partir da visão apical esquerda de 4 câmaras otimizada para o ventrículo direito
Para os grupos 2-5, o tratamento deve ser orientado para o tratamento da condição subjacente. Além disso, deve concentrar-se em manter/melhorar a oxigenação. Os pacientes devem ser encaminhados para especialistas em centro hospitalar terciário para o tratamento, que é muitas vezes complexo.
Agentes vasodilatadores
Bloqueadores dos canais de cálcio (BCCs):
Só é eficaz em pacientes com vasorreatividade
Os BCCs bloqueiam a entrada de cálcio:
Nas células endoteliais → vasodilatação
Nos nós sinusal (SA) e auriculoventricular (AV) → ↓ condução cardíaca e contratilidade
Dihidropiridínicos:
Principalmente vasodilatadores em dose terapêutica
Doses elevadas podem produzir uma redução dramática da pressão da artéria pulmonar.
Prostaglandinas vasodilatadoras:
A prostaciclina (PGIPGIAn aldose-ketose isomerase that catalyzes the reversible interconversion of glucose 6-phosphate and fructose 6-phosphate. In prokaryotic and eukaryotic organisms it plays an essential role in glycolytic and gluconeogenic pathways. In mammalian systems the enzyme is found in the cytoplasm and as a secreted protein. This secreted form of glucose-6-phosphate isomerase has been referred to as autocrine motility factor or neuroleukin, and acts as a cytokine which binds to the autocrine motility factor receptor. Deficiency of the enzyme in humans is an autosomal recessive trait, which results in congenital nonspherocytic hemolytic anemia.Glycolysis2) é uma substância vasodilatadora natural.
Análogos PGIPGIAn aldose-ketose isomerase that catalyzes the reversible interconversion of glucose 6-phosphate and fructose 6-phosphate. In prokaryotic and eukaryotic organisms it plays an essential role in glycolytic and gluconeogenic pathways. In mammalian systems the enzyme is found in the cytoplasm and as a secreted protein. This secreted form of glucose-6-phosphate isomerase has been referred to as autocrine motility factor or neuroleukin, and acts as a cytokine which binds to the autocrine motility factor receptor. Deficiency of the enzyme in humans is an autosomal recessive trait, which results in congenital nonspherocytic hemolytic anemia.Glycolysis2: epoprostenolEpoprostenolA prostaglandin that is a powerful vasodilator and inhibits platelet aggregation. It is biosynthesized enzymatically from prostaglandin endoperoxides in human vascular tissue. The sodium salt has been also used to treat primary pulmonary hypertension.Hemostasis, treprostinilTreprostinilPulmonary Hypertension Drugs
Antagonistas dos recetores da endotelina:
As endotelinas são vasoconstritores naturais.
Antagonizam competitivamente os recetores de endotelina → ↓ resistência vascular pulmonar
Exemplos: sildenafilSildenafilA phosphodiesterase type-5 inhibitor; vasodilator agent and urological agent that is used in the treatment of erectile dysfunction and primary pulmonary hypertension.Phosphodiesterase Inhibitors (Viagra), tadalafilTadalafilA carboline derivative and phosphodiesterase 5 inhibitor that is used primarily to treat erectile dysfunction; benign prostatic hyperplasia and primary pulmonary hypertension.Phosphodiesterase Inhibitors
Ativadores da guanilato ciclase
A guanilato ciclase solúvel (sGC, pela sigla em inglês) é um recetor intracelular de NO.
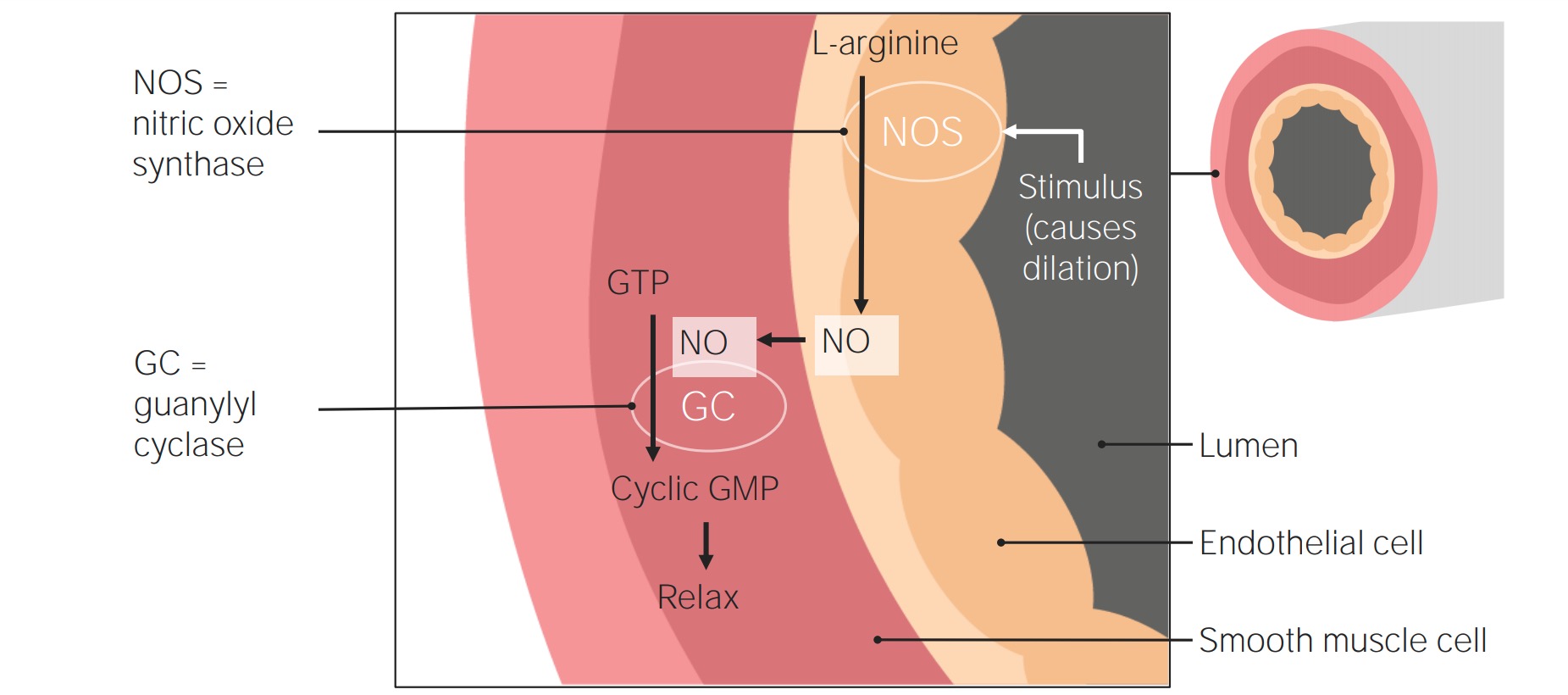
Agentes vasodilatadores no tratamento da hipertensão pulmonar: Um estímulo ativa a NO sintase (NOS) nas células endoteliais para converter a L-arginina em NO. O NO move-se para o músculo liso, onde estimula a atividade da guanilato ciclase (também conhecida como guanilil ciclase), que converte o trifosfato de guanosina em cGMP. O GMP cíclico induz então um relaxamento do músculo liso, provocando vasodilatação.
Imagem por Lecturio.
Outras opções de tratamento não cirúrgico
Medidas de suporte:
Oxigenoterapia
Dieta hipossalina
Fisioterapia/exercício supervisionado para melhorar a capacidade funcional
Vacinação contra a gripe
Controlo da natalidade para prevenir a gravidez devido ao elevado risco de mortalidade materna associada à HP na gravidez
Diuréticos:
Indicação: pacientes com sobrecarga de volume do lado direito (edemaEdemaEdema is a condition in which excess serous fluid accumulates in the body cavity or interstitial space of connective tissues. Edema is a symptom observed in several medical conditions. It can be categorized into 2 types, namely, peripheral (in the extremities) and internal (in an organ or body cavity). Edema, ascite)
Evitar hipovolemia (os pacientes com insuficiência cardíaca direita são dependentes da pré-carga).
Os diuréticos da ansa são normalmente os fármacos de 1ª linha (por exemplo, furosemida).
Anticoagulação:
Indicações:
Grupo 4 (HP por doença tromboembólica crónica)
HAP do grupo 1 idiopática ou familiar
Fibrilhação auricular
A varfarina é normalmente o fármaco de eleição.
Digoxina:
Indicações:
Insuficiência cardíaca devido a disfunção sistólica
Algumas taquiarritmias supraventriculares
Efeitos:
Inotrópico positivo
Pode reduzir a ativação simpática
Opções cirúrgicas
Transplante pulmonar:
Indicações para referenciação:
Doença rapidamente progressiva, já sob terapêutica
Uso de prostaglandinas por via parentérica
Doença veno-oclusiva pulmonar conhecida ou suspeita
Procedimentos de eleição:
Transplante pulmonar bilateral
Transplante de coração-pulmão
Criação de um shunt direito-esquerdo
Pode ser usado como ponte para transplante ou como paliativo
Procedimentos:
Septostomia auricular
Shunt de Potts, por cateter: colocação de um shunt entre a artéria pulmonar esquerda e a aortaAortaThe main trunk of the systemic arteries.Mediastinum and Great Vessels: Anatomy descendente
Trombectomia endovascular: em pacientes com tromboembolia crónica e com fonte conhecida
Prognóstico
A HAP é progressiva e pode ser fatal se não tratada.
Sem tratamento, a HAP do grupo 1 tem o pior prognóstico:
Sobrevivência a 1 ano: 85%
Sobrevivência a 3 anos: 68%
Sobrevivência a 5 anos: 57%
O prognóstico depende da causa subjacente.
A principal causa de morte é falência ventricular direita.
Os pacientes devem ser monitorizados por um especialista para avaliar se há diminuição do estado funcional.
Insuficiência cardíaca: incapacidade do coração fornecer um débito cardíaco normal ao organismo para satisfazer as necessidades metabólicas. A insuficiência cardíaca pode levar a HP do grupo 2. O ecocardiograma pode confirmar o diagnóstico e fornecer informações sobre a fração de ejeção. O tratamento baseia-se na remoção do excesso de líquidos e diminuição das necessidades de oxigénio do coração.
Doença das artérias coronárias: devido a estenose das artérias coronárias, levando a isquemia cardíaca. Os sintomas incluem toracalgia e dispneia. O diagnóstico baseia-se na história, achados do ECGECGAn electrocardiogram (ECG) is a graphic representation of the electrical activity of the heart plotted against time. Adhesive electrodes are affixed to the skin surface allowing measurement of cardiac impulses from many angles. The ECG provides 3-dimensional information about the conduction system of the heart, the myocardium, and other cardiac structures. Electrocardiogram (ECG), testesTestesGonadal Hormones de stress cardíaco e/ou cateterismo cardíaco. O tratamento passa pela redução das necessidades de oxigénio do coração e no aumento do fornecimento de oxigénio.
Insuficiência tricúspide: defeito valvular que permite o refluxo de sangue do ventrículo direito para a aurícula direita durante a sístole.A insuficiência tricúspide pode ser assintomática ou associar-se a congestão venosa sistémica devido ao aumento das pressões auriculares e venosas direitas. Também pode levar a HP, mas a doença valvular intrínseca deve ser considerada no diagnóstico diferencial da HP. A ecocardiografia pode ajudar a estabelecer o diagnóstico. O tratamento concentra-se no controlo da insuficiência cardíaca, e a cirurgia está reservada para casos graves.
Fibrose pulmonar: uma doença pulmonar intersticial rapidamente progressiva com poucos tratamentos disponíveis. À medida que a fibrose pulmonar progride, pode desenvolver-se HP do grupo 3. A esperança média de vida é de 3-4 anos desde o diagnóstico. O transplante pulmonar é a única intervenção curativa, desde que cumpra os seus critérios.
DPOC: doença pulmonar geralmente causada pelo tabagismo e caracterizada por obstrução progressiva e irreversível do fluxo aéreo secundária à inflamação crónica. A doença pulmonar obstrutiva crónica é uma das principais causas da HP do grupo 3 e os sintomas incluem dispneia progressiva e tosse crónica. O diagnóstico é confirmado com testesTestesGonadal Hormones de função pulmonar. O tratamento inclui cessação tabágica, reabilitação pulmonar e farmacoterapia.
Embolia pulmonar: resulta da obstrução intraluminal da artéria pulmonar principal ou dos seus ramos por certos componentes (por exemplo, trombo, colesterol, arARAortic regurgitation (AR) is a cardiac condition characterized by the backflow of blood from the aorta to the left ventricle during diastole. Aortic regurgitation is associated with an abnormal aortic valve and/or aortic root stemming from multiple causes, commonly rheumatic heart disease as well as congenital and degenerative valvular disorders. Aortic Regurgitation, líquido amniótico ou gordura). O sintoma maisMAISAndrogen Insensitivity Syndrome comum é a dispneia. O tratamento inicial é de suporte (com foco no restabelecimento da oxigenação e estabilidade hemodinâmica), seguido de anticoagulação sistémica e terapias de intervenção. Os eventos tromboembólicos crónicos podem levar a HP do grupo 4.
Simonneau, G., Gatzoulis, M.A., Adatia, I., Celermajer, D., Denton, C., Ghofrani, A., Gomez Sanchez, M.A., Krishna Kumar, R., Landzberg, M., Machado, R.F., Olschewski, H., Robbins, I.M., Souza, R. (2013). Updated clinical classification of pulmonary hypertension. Journal of the American College of Cardiology, 62(25 Suppl), D34–D41. https://doi.org/10.1016/j.jacc.2013.10.029
A Lecturio Medical complementa o teu estudo através de métodos de ensino baseados em evidência, vídeos de palestras, perguntas e muito mais – tudo combinado num só lugar e fácil de usar.
User Reviews
Details
×
Obtenha Premium para testar os seus conhecimentos
Lecturio Premium dá-lhe acesso total a todos os conteúdos e características
Obtenha Premium para ver todos os vídeos
Verifique agora o seu e-mail para obter um teste gratuito.
Crie uma conta gratuita para testar os seus conhecimentos
Lecturio Premium dá-lhe acesso total a todos os conteúdos e características - incluindo o Qbank de Lecturio com perguntas actualizadas ao estilo do board-.