


A hiperplasia adrenal congénita (HAC) consiste num grupo de doenças autossómicas recessivas que causam défice de uma enzima necessária à síntese de cortisol Cortisol Glucocorticoids, de aldosterona e de androgénios. A subforma mais MAIS Androgen Insensitivity Syndrome comum de HAC é o défice de 21-hidroxilase, seguido pelo défice de 11β-hidroxilase. As manifestações clínicas dependem da enzima específica afetada. A HAC é a causa mais MAIS Androgen Insensitivity Syndrome comum de genitália ambígua em indivíduos com genótipo feminino. Todas as formas de HAC causam baixos níveis de cortisol Cortisol Glucocorticoids, altos níveis de hormona adrenocorticotrófica (ACTH) e hiperplasia adrenal. Os exames laboratoriais ajudam a confirmar o diagnóstico. É necessário fazer-se reposição de glucocorticoides durante toda a vida, e a correção cirúrgica de genitais ambíguos é realizada frequentemente.
Last updated: Dec 15, 2025
A hiperplasia adrenal congénita compreende um espectro de perturbações em que diferentes mutações genéticas causam diferentes défices enzimáticos que afetam a biogénese de esteroides, que é realizada pelas glândulas adrenais.
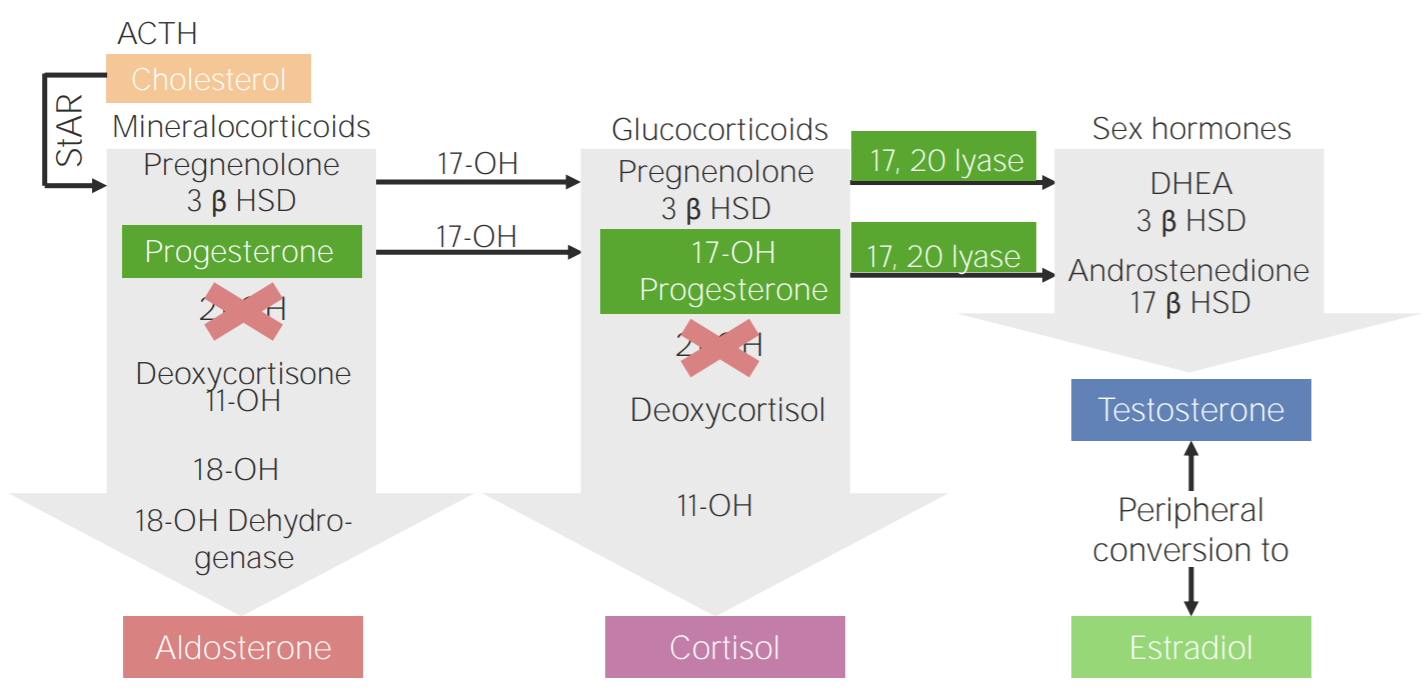
Défice de 21-hidroxilase: diagrama da fisiopatologia da hiperplasia adrenal congénita devido ao défice de 21-hidroxilase
3β HSD: 3β-hidroxiesteroide desidrogenase
11-OH: 11β-hidroxilase
17-OH: 17α-hidroxilase
17-OH progesterone: 17-hidroxiprogesterona
17β HSD: 17β-hidroxiesteroide-desidrogenase
18-OH: 18-hidroxilase
21-OH: 21-hidroxilase
ACTH, pela sigla em inglês: hormona adrenocorticotrópica
DHEA: dehidroepiandrosterona
StAR, pela sigla em inglês: proteína reguladora aguda esteroidogénica
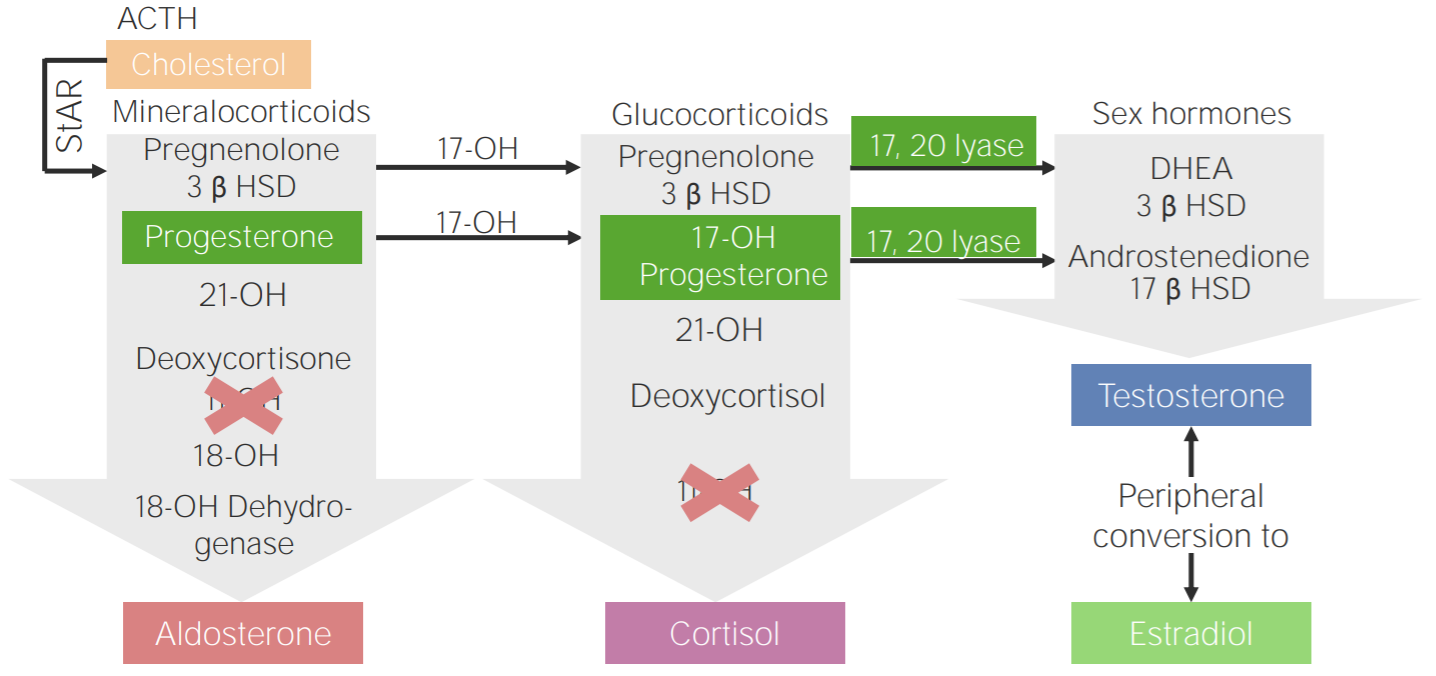
Défice de 11β-hidroxilase: diagrama da fisiopatologia da hiperplasia adrenal congénita devido ao défice de 11β-hidroxilase
3β HSD: 3β-hidroxiesteroide desidrogenase
11-OH: 11β-hidroxilase
17-OH: 17α-hidroxilase
17-OH progesterone: 17-hidroxiprogesterona
17β HSD: 17β-hidroxiesteroide-desidrogenase
18-OH: 18-hidroxilase
21-OH: 21-hidroxilase
ACTH, pela sigla em inglês: hormona adrenocorticotrópica
DHEA: dehidroepiandrosterona
StAR, pela sigla em inglês: proteína reguladora aguda esteroidogénica
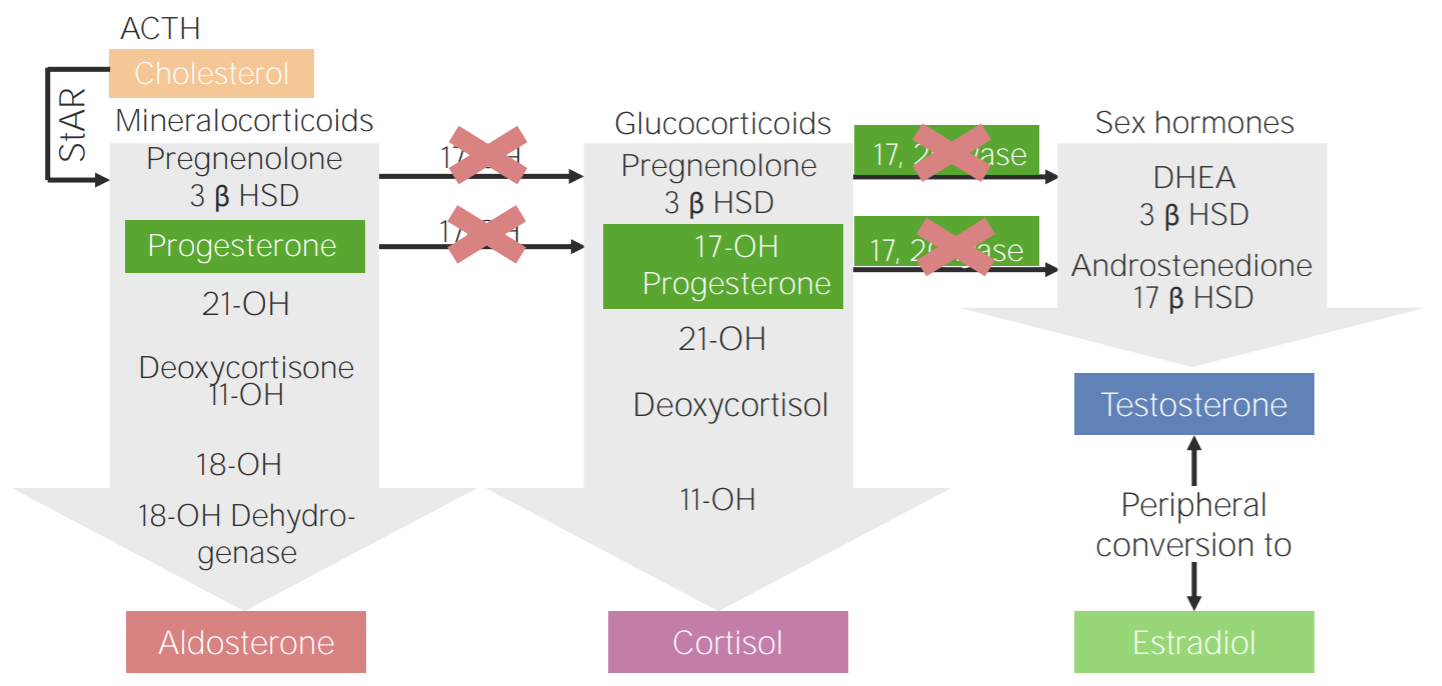
Défice de 17α-hidroxilase: diagrama da fisiopatologia da hiperplasia adrenal congénita devido ao défice de 17α-hidroxilase
3β HSD: 3β-hidroxiesteroide desidrogenase
11-OH: 11β-hidroxilase
17-OH: 17α-hidroxilase
17-OH progesterone: 17-hidroxiprogesterona
17β HSD: 17β-hidroxiesteroide-desidrogenase
18-OH: 18-hidroxilase
21-OH: 21-hidroxilase
ACTH, pela sigla em inglês: hormona adrenocorticotrópica
DHEA: dehidroepiandrosterona
StAR, pela sigla em inglês: proteína reguladora aguda esteroidogénica
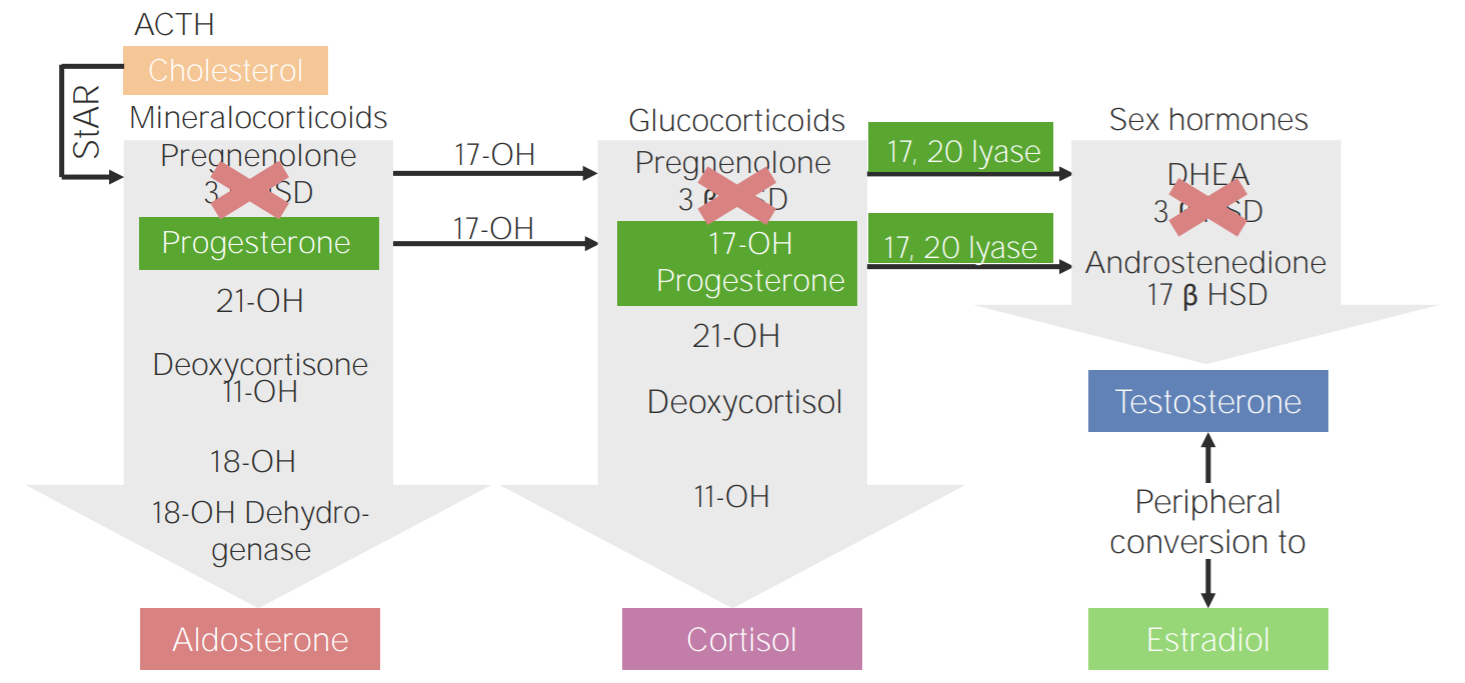
Défice de 3β-hidroxiesteroide desidrogenase: diagrama da fisiopatologia da hiperplasia adrenal congénita devido ao défice de 3β-hidroxiesteroide desidrogenase
3β HSD: 3β-hidroxiesteroide desidrogenase
11-OH: 11β-hidroxilase
17-OH: 17α-hidroxilase
17-OH progesterone: 17-hidroxiprogesterona
17β HSD: 17β-hidroxiesteroide-desidrogenase
18-OH: 18-hidroxilase
21-OH: 21-hidroxilase
ACTH, pela sigla em inglês: hormona adrenocorticotrópica
DHEA: dehidroepiandrosterona
StAR, pela sigla em inglês: proteína reguladora aguda esteroidogénica
| Défice de enzima | Gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics mutado | Cortisol Cortisol Glucocorticoids | Aldosterona | 11-desoxicorticosterona | Androgénios |
|---|---|---|---|---|---|
| 21-hidroxilase | CYP21A2 | Diminuida | Diminuida | Diminuida | Aumentados |
| 11β-hidroxilase | CYP11B1 | Aumentada | |||
| 17α-hidroxilase | CYP17A1 | Diminuidos | |||
| 3BHSD 3BHSD Congenital Adrenal Hyperplasia | HSD3B2 | Diminuida |
Existe uma forma clássica de HAC devido ao défice de 21-hidroxilase e uma forma não clássica. A forma clássica tem 2 tipos diferentes: virilizante simples e perdedora de sal (salt-wasting).
A HAC clássica é a forma mais MAIS Androgen Insensitivity Syndrome grave de défice de 21-hidroxilase.
A HAC não clássica é a forma mais MAIS Androgen Insensitivity Syndrome ligeira e comum de défice de 21-hidroxilase.
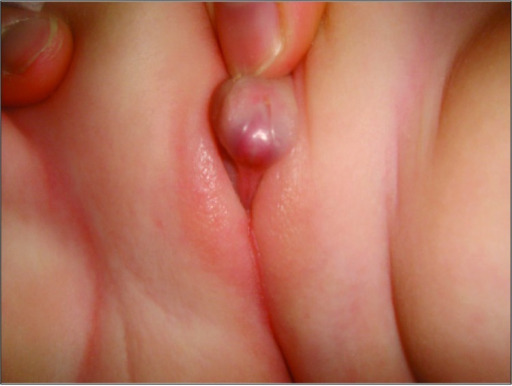
Aparência de genitália ambígua num paciente com hiperplasia adrenal congénita
Imagem: “A rare combination: congenital adrenal hyperplasia due to 21 hydroxylase deficiency and Turner syndrome” por Kendirci HN, Aycan Z, Çetinkaya S, Baş VN, Ağladıoğlu SY, Önder A. License: CC BY 2.5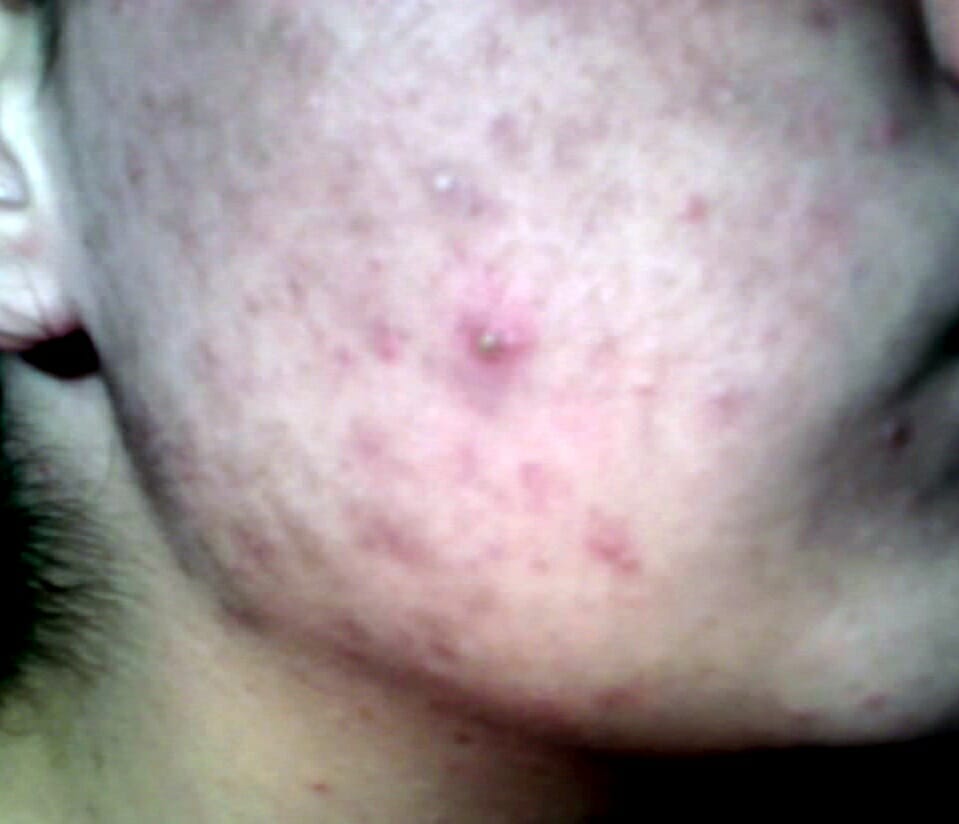
Acne na face de uma rapariga durante a puberdade: mais visto na hiperplasia adrenal congénita não-clássica, uma forma comum de défice de 21-alfa-hidroxilase
Imagem: “Teenager-with-acne” por Diariodaj. Licença: Public DomainA tabela a seguir descreve achados importantes nos 3 tipos mais MAIS Androgen Insensitivity Syndrome comuns de HAC.
| Tipo de HAC | Esteroides/precursores | Eletrólitos |
|---|---|---|
| 21-hidroxilase |
|
|
| 11β-hidroxilase |
|
|
| 17α-hidroxilase |
|
Os objetivos gerais do tratamento são mitigar os efeitos dos respetivos défices de esteroides e quantidades excessivas de esteroides e ajudar no desenvolvimento sexual secundário.
