La hiperplasia suprarrenal congénita (HSC) consiste en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum un grupo de trastornos autosómicos recesivos que causan una deficiencia de una enzima necesaria para la síntesis de cortisol Cortisol Glucocorticoids, aldosterona y andrógenos. La subforma más común de HSC es la deficiencia de 21-hidroxilasa, seguida de la de 11β-hidroxilasa. Las manifestaciones clínicas dependen de la enzima específica afectada. En EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum particular, la HSC es la causa más común de genitales ambiguos en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum individuos genotípicamente femeninos. Todas las formas de HSR causan niveles bajos de cortisol Cortisol Glucocorticoids, niveles altos de la hormona adrenocorticotrópica (ACTH) e hiperplasia suprarrenal. Los LOS Neisseria estudios de laboratorio ayudan a confirmar el diagnóstico. Es necesario el reemplazo de glucocorticoides de por vida, y a menudo se realiza la corrección quirúrgica de los LOS Neisseria genitales ambiguos.
Last updated: Dec 15, 2025
La hiperplasia suprarrenal congénita comprende un espectro de trastornos en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria que diferentes mutaciones genéticas causan distintas deficiencias enzimáticas que afectan a la biogénesis de los LOS Neisseria esteroides, de la que se encargan las glándulas suprarrenales.
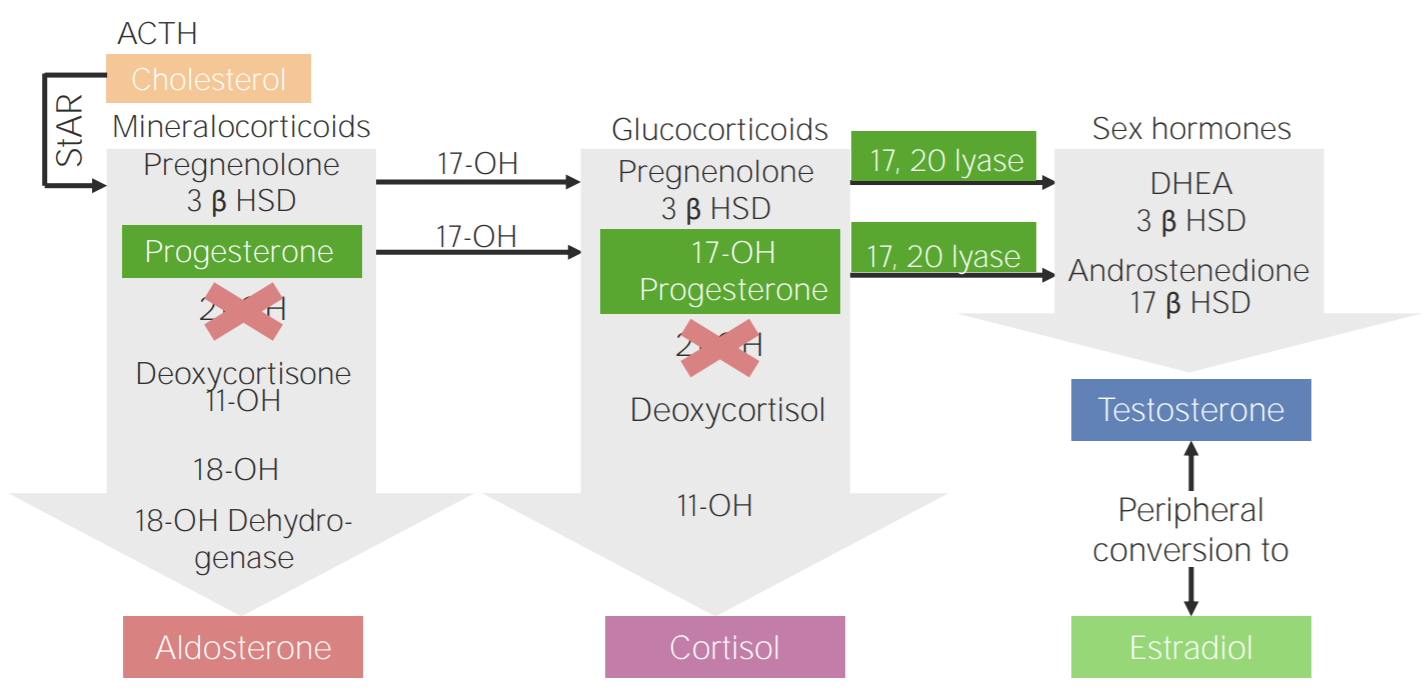
Deficiencia de 21-hidroxilasa: esquema de la fisiopatología de la hiperplasia suprarrenal congénita por deficiencia de 21-hidroxilasa
3β HSD: 3β-hidroxiesteroide deshidrogenasa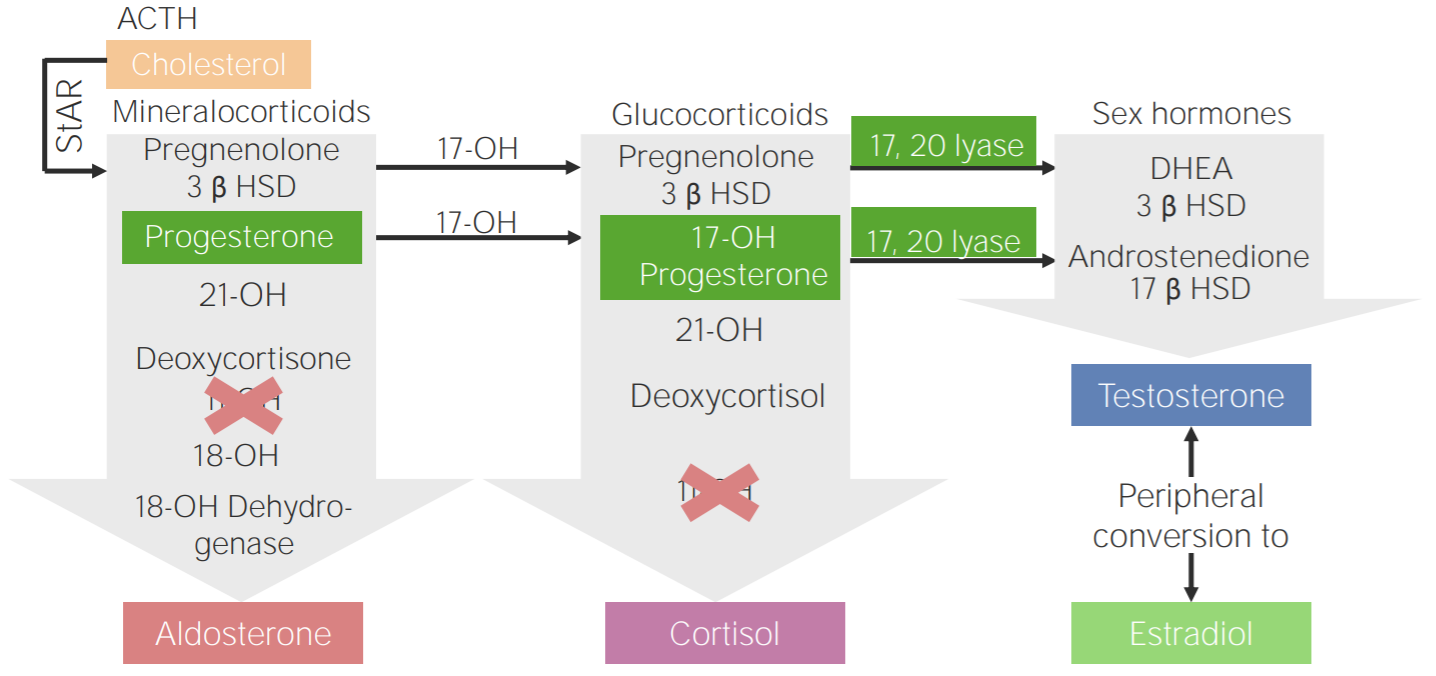
Deficiencia de 11β-hidroxilasa: esquema de la fisiopatología de la hiperplasia suprarrenal congénita por deficiencia de 11β-hidroxilasa
3β HSD: 3β-hidroxiesteroide deshidrogenasa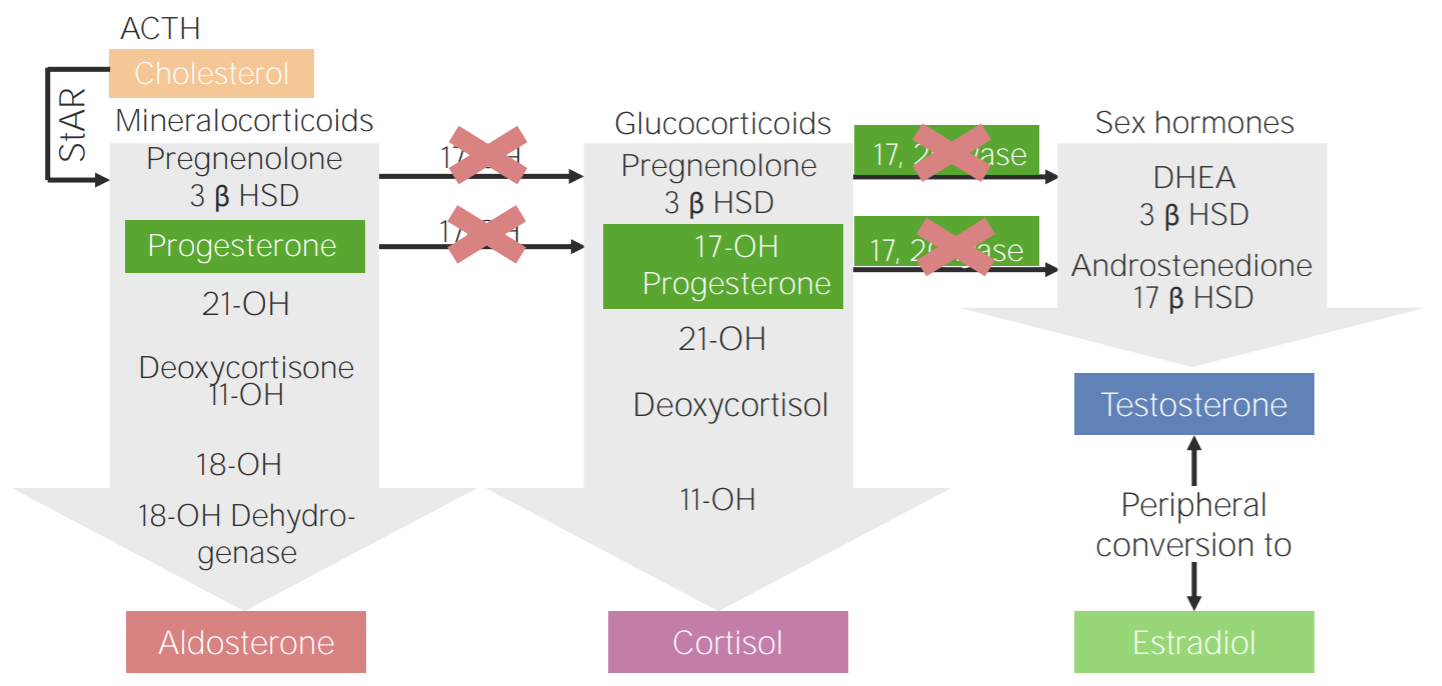
Deficiencia de 17α-hidroxilasa: esquema de la fisiopatología de la hiperplasia suprarrenal congénita por deficiencia de 17α-hidroxilasa
3β HSD: 3β-hidroxiesteroide deshidrogenasa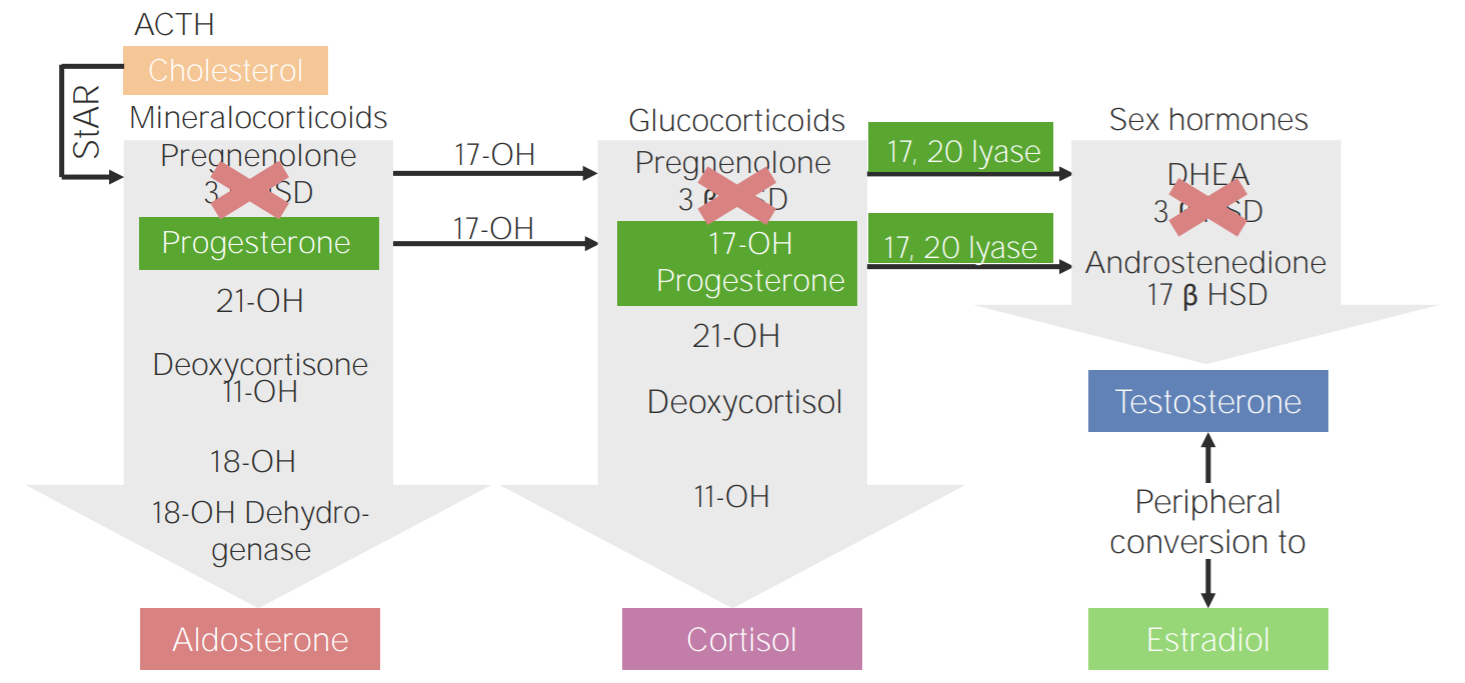
Deficiencia de 3β-hidroxiesteroide deshidrogenasa: esquema de la fisiopatología de la hiperplasia suprarrenal congénita por deficiencia de 3β-hidroxiesteroide deshidrogenasa
3β HSD: 3β-hidroxiesteroide deshidrogenasa| Deficiencia enzimática | Gen mutado | Cortisol Cortisol Glucocorticoids | Aldosterona | 11-deoxicorticosterona | Andrógenos |
|---|---|---|---|---|---|
| 21-hidroxilasa | CYP21A2 | Disminuida | Disminuida | Disminuida | Aumentada |
| 11β-hidroxilasa | CYP11B1 | Aumentada | |||
| 17α-hidroxilasa | CYP17A1 | Disminuida | |||
| 3BHSD 3BHSD Congenital Adrenal Hyperplasia | HSD3B2 | Disminuida |
Existe una forma clásica de HSC debida al AL Amyloidosis déficit de 21-hidroxilasa y una forma no clásica. La forma clásica tiene 2 tipos diferentes: la virilizante simple y la de desgaste de sal.
La HSC clásica es la forma más grave de deficiencia de 21-hidroxilasa.
La HSC no clásica es la forma más leve y común de la deficiencia de 21-hidroxilasa.
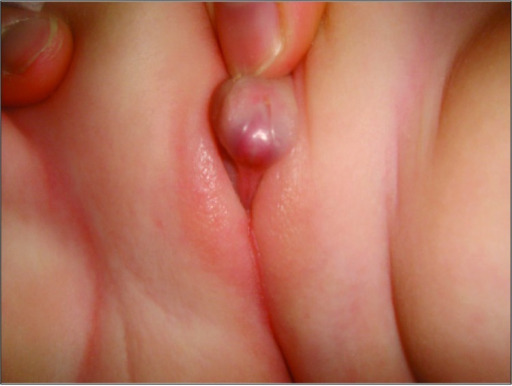
Aparición de genitales ambiguos en un paciente con hiperplasia suprarrenal congénita
Imagen: “A rare combination: congenital adrenal hyperplasia due to 21 hydroxylase deficiency and Turner syndrome” by Kendirci HN, Aycan Z, Çetinkaya S, Baş VN, Ağladıoğlu SY, Önder A. License: CC BY 2.5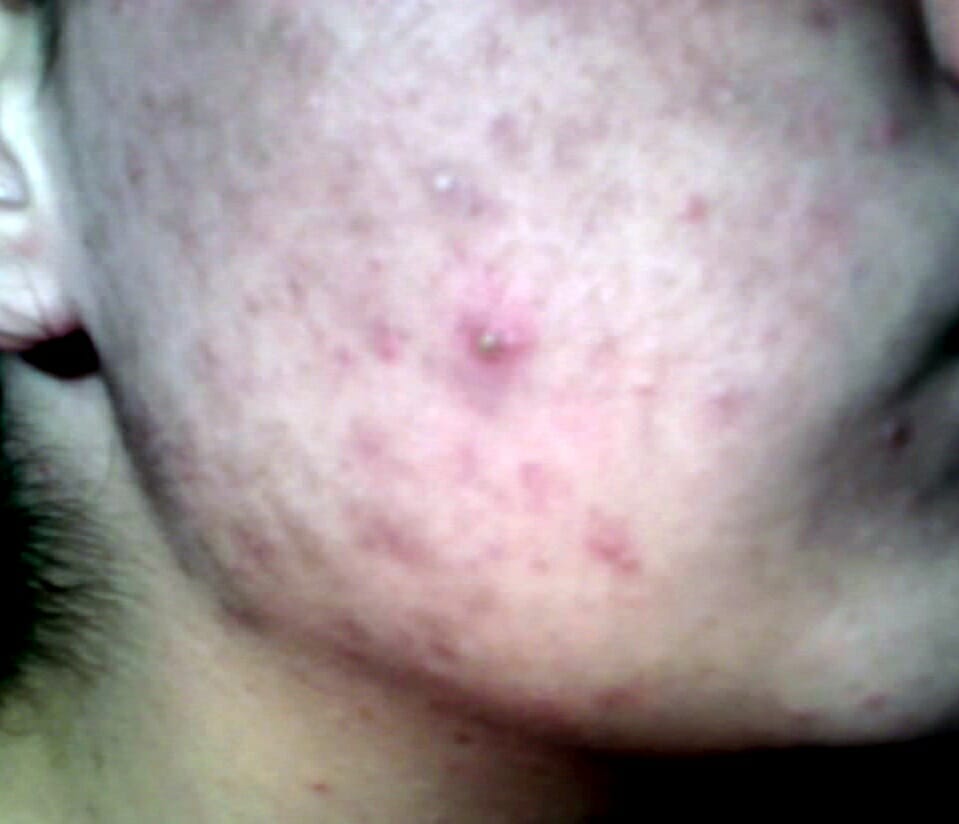
Acné en la frente de una niña durante la pubertad: Se observa en la hiperplasia suprarrenal congénita no clásica de forma más frecuente, una forma común de deficiencia de 21-alfa-hidroxilasa.
Imagen: “Teenager-with-acne” por Diariodaj. Licencia: Dominio PúblicoEn EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la siguiente tabla se describen los LOS Neisseria hallazgos importantes en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria 3 tipos más comunes de HSC.
| Tipo de HSC | Esteroides/precursores | Electrolitos |
|---|---|---|
| 21-hidroxilasa |
|
|
| 11β-hidroxilasa |
|
|
| 17α-hidroxilasa |
|
Los LOS Neisseria objetivos generales del tratamiento son mitigar los LOS Neisseria efectos de las respectivas deficiencias de esteroides y de las cantidades excesivas de los LOS Neisseria mismos y ayudar al AL Amyloidosis desarrollo sexual secundario.