


A galactosemia Galactosemia Galactosemia is a disorder caused by defects in galactose metabolism. Galactosemia is an inherited, autosomal-recessive condition, which results in inadequate galactose processing and high blood levels of monosaccharide. The rare disorder often presents in infants with symptoms of lethargy, nausea, vomiting, diarrhea, and jaundice. Galactosemia é uma doença causada por defeitos no metabolismo da galactose Galactose An aldohexose that occurs naturally in the d-form in lactose, cerebrosides, gangliosides, and mucoproteins. Deficiency of galactosyl-1-phosphate uridyltransferase causes an error in galactose metabolism called galactosemia, resulting in elevations of galactose in the blood. Lactose Intolerance. A galactosemia Galactosemia Galactosemia is a disorder caused by defects in galactose metabolism. Galactosemia is an inherited, autosomal-recessive condition, which results in inadequate galactose processing and high blood levels of monosaccharide. The rare disorder often presents in infants with symptoms of lethargy, nausea, vomiting, diarrhea, and jaundice. Galactosemia é uma condição herdada, autossómica recessiva, que resulta num processamento inadequado da galactose Galactose An aldohexose that occurs naturally in the d-form in lactose, cerebrosides, gangliosides, and mucoproteins. Deficiency of galactosyl-1-phosphate uridyltransferase causes an error in galactose metabolism called galactosemia, resulting in elevations of galactose in the blood. Lactose Intolerance e em níveis elevados do monossacarídeo no sangue. A doença rara apresenta-se frequentemente em bebés com sintomas de letargia, náuseas, vómitos, diarreia e icterícia. Podem ocorrer complicações neurológicas sérias, como deficiências motoras (por exemplo, ataxia Ataxia Impairment of the ability to perform smoothly coordinated voluntary movements. This condition may affect the limbs, trunk, eyes, pharynx, larynx, and other structures. Ataxia may result from impaired sensory or motor function. Sensory ataxia may result from posterior column injury or peripheral nerve diseases. Motor ataxia may be associated with cerebellar diseases; cerebral cortex diseases; thalamic diseases; basal ganglia diseases; injury to the red nucleus; and other conditions. Ataxia-telangiectasia) e na fala. O diagnóstico é feito através de testes Testes Gonadal Hormones de sangue, que detetam a ausência ou o nível baixo das enzimas necessárias para processar a galactose Galactose An aldohexose that occurs naturally in the d-form in lactose, cerebrosides, gangliosides, and mucoproteins. Deficiency of galactosyl-1-phosphate uridyltransferase causes an error in galactose metabolism called galactosemia, resulting in elevations of galactose in the blood. Lactose Intolerance. O tratamento consiste em evitar a lactose e a galactose Galactose An aldohexose that occurs naturally in the d-form in lactose, cerebrosides, gangliosides, and mucoproteins. Deficiency of galactosyl-1-phosphate uridyltransferase causes an error in galactose metabolism called galactosemia, resulting in elevations of galactose in the blood. Lactose Intolerance na alimentação.
Last updated: Apr 17, 2025
A galactosemia Galactosemia Galactosemia is a disorder caused by defects in galactose metabolism. Galactosemia is an inherited, autosomal-recessive condition, which results in inadequate galactose processing and high blood levels of monosaccharide. The rare disorder often presents in infants with symptoms of lethargy, nausea, vomiting, diarrhea, and jaundice. Galactosemia é um erro autossómico recessivo e inato do metabolismo dos carboidratos, que resulta na incapacidade do organismo de metabolizar a galactose Galactose An aldohexose that occurs naturally in the d-form in lactose, cerebrosides, gangliosides, and mucoproteins. Deficiency of galactosyl-1-phosphate uridyltransferase causes an error in galactose metabolism called galactosemia, resulting in elevations of galactose in the blood. Lactose Intolerance em glicose.
A galactosemia Galactosemia Galactosemia is a disorder caused by defects in galactose metabolism. Galactosemia is an inherited, autosomal-recessive condition, which results in inadequate galactose processing and high blood levels of monosaccharide. The rare disorder often presents in infants with symptoms of lethargy, nausea, vomiting, diarrhea, and jaundice. Galactosemia é uma doenla hereditária causada por uma mutação genética, que leva à deficiência enzimática.
A patologia é autossómica recessiva: Cada criança tem 25% de probabilidade de herdar a doença se ambos os pais PAIS Androgen Insensitivity Syndrome estão afetados.
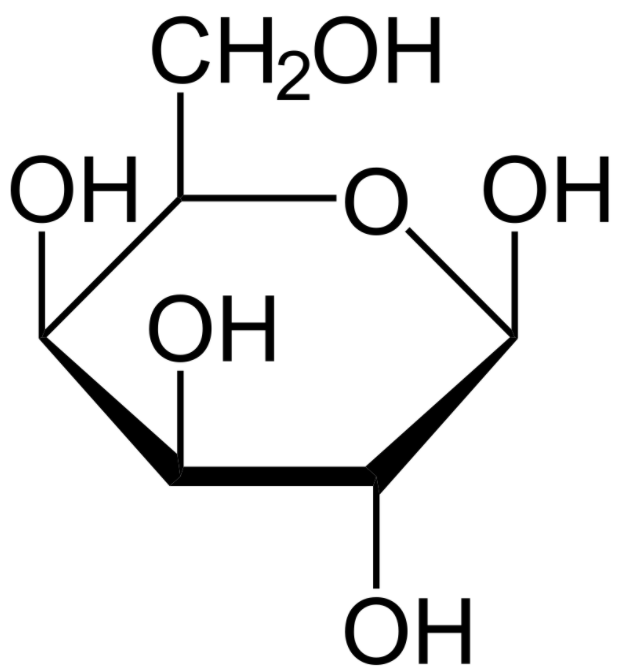
Uma estrutura do monossacarídeo galactose (projeção de Haworth):
Os pacientes com galactosemia são incapazes de degradar a galactose. A glicose e a galactose têm a mesma fórmula química, mas diferem na localização do grupo hidroxil 1.
Os sintomas podem aparecer dias a semanas após o nascimento.
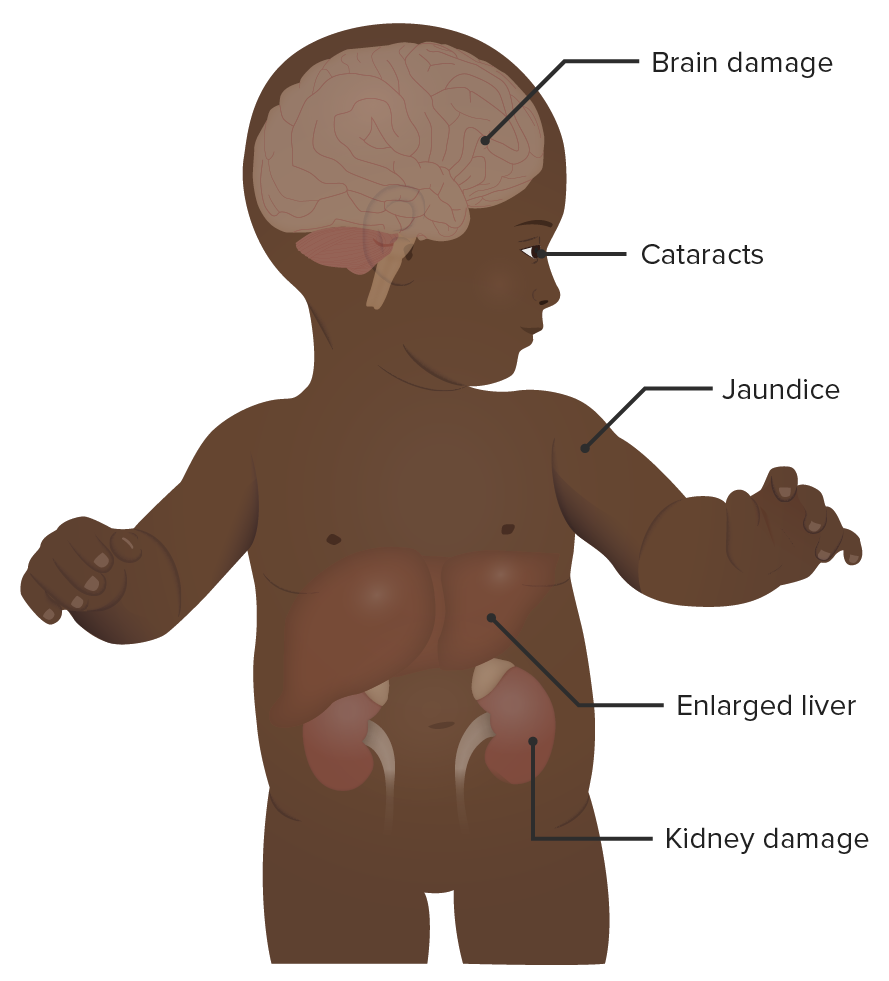
Um bebé com galactosemia:
Como se observa na imagem, a galactosemia tem vários sinais e sintomas clínicos. Podem ocorrer efeitos secundários neurológicos, renais, hepáticos e oculares. Os sintomas aparecem cedo e são frequentemente aparentes no 1.º mês de vida.
O teste deve ser realizado em todos os bebés que apresentem sintomas.
