La galactosemia Galactosemia Galactosemia is a disorder caused by defects in galactose metabolism. Galactosemia is an inherited, autosomal-recessive condition, which results in inadequate galactose processing and high blood levels of monosaccharide. The rare disorder often presents in infants with symptoms of lethargy, nausea, vomiting, diarrhea, and jaundice. Galactosemia es un trastorno causado por defectos en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el metabolismo de la galactosa. La galactosemia Galactosemia Galactosemia is a disorder caused by defects in galactose metabolism. Galactosemia is an inherited, autosomal-recessive condition, which results in inadequate galactose processing and high blood levels of monosaccharide. The rare disorder often presents in infants with symptoms of lethargy, nausea, vomiting, diarrhea, and jaundice. Galactosemia es una condición hereditaria, autosómica recesiva, que resulta en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum un procesamiento inadecuado de la galactosa y niveles altos de monosacáridos en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la sangre. El raro trastorno a menudo se presenta en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum bebés con síntomas de letargo, náuseas, vómitos, diarrea e ictericia. Pueden ocurrir complicaciones neurológicas graves, como deficiencias motoras y del habla (e.g., ataxia Ataxia Impairment of the ability to perform smoothly coordinated voluntary movements. This condition may affect the limbs, trunk, eyes, pharynx, larynx, and other structures. Ataxia may result from impaired sensory or motor function. Sensory ataxia may result from posterior column injury or peripheral nerve diseases. Motor ataxia may be associated with cerebellar diseases; cerebral cortex diseases; thalamic diseases; basal ganglia diseases; injury to the red nucleus; and other conditions. Ataxia-telangiectasia). El diagnóstico se realiza mediante un análisis de sangre, que detecta la ausencia o el bajo nivel de las enzimas necesarias para procesar la galactosa. El tratamiento consiste en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum evitar la lactosa y la galactosa en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la dieta.
Last updated: Apr 17, 2025
La galactosemia Galactosemia Galactosemia is a disorder caused by defects in galactose metabolism. Galactosemia is an inherited, autosomal-recessive condition, which results in inadequate galactose processing and high blood levels of monosaccharide. The rare disorder often presents in infants with symptoms of lethargy, nausea, vomiting, diarrhea, and jaundice. Galactosemia es un error Error Refers to any act of commission (doing something wrong) or omission (failing to do something right) that exposes patients to potentially hazardous situations. Disclosure of Information congénito autosómico recesivo del metabolismo de los LOS Neisseria carbohidratos que resulta en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la incapacidad del cuerpo para metabolizar la galactosa en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum glucosa.
La galactosemia Galactosemia Galactosemia is a disorder caused by defects in galactose metabolism. Galactosemia is an inherited, autosomal-recessive condition, which results in inadequate galactose processing and high blood levels of monosaccharide. The rare disorder often presents in infants with symptoms of lethargy, nausea, vomiting, diarrhea, and jaundice. Galactosemia es un trastorno hereditario causado por una mutación genética que conduce a una deficiencia enzimática.
La condición es autosómica recesiva: cada hijo tiene un 25 % de posibilidades de heredar la enfermedad si ambos padres están afectados.
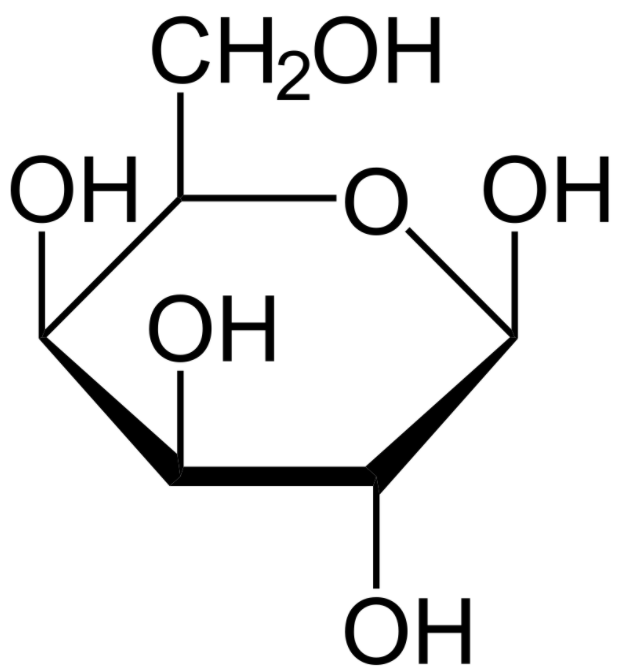
Una estructura del monosacárido galactosa (proyección de Haworth):
Los pacientes con galactosemia no pueden degradar la galactosa. La glucosa y la galactosa tienen la misma fórmula química, pero difieren en la ubicación del grupo 1 hidroxilo.
Los LOS Neisseria síntomas pueden aparecer días o semanas después del nacimiento.
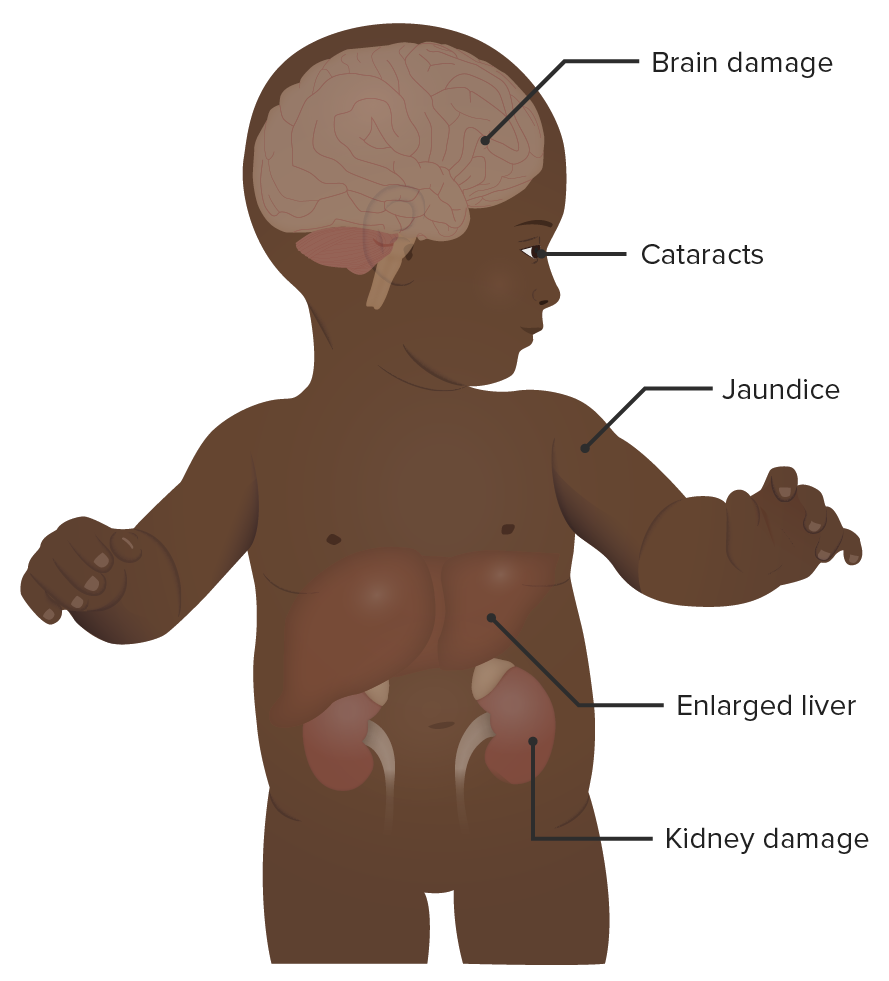
Un bebé con galactosemia:
Como se observa en la imagen, la galactosemia tiene numerosos signos y síntomas clínicos. Pueden ocurrir efectos secundarios neurológicos, renales, hepáticos y oculares. Los síntomas aparecen temprano y, a menudo, son evidentes dentro del primer mes de vida.
La prueba debe realizarse en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum todos los LOS Neisseria bebés que presenten síntomas.