


A farmacocinética é a ciência que analisa como o corpo humano interage com um fármaco. A farmacocinética examina como é que o fármaco é absorvido, distribuído, metabolizado e excretado pelo organismo. A farmacodinâmica é a ciência que estuda os efeitos bioquímicos e fisiológicos de um fármaco e o seu mecanismo de ação órgão-específico, incluindo efeitos ao nível celular. Outra maneira de descrever a diferença entre as duas disciplinas é dizer que a farmacocinética é “o que o corpo faz com o fármaco”, enquanto a farmacodinâmica é “o que o fármaco faz com o corpo”. Ao prescrever a terapêutica, os médicos devem ter em consideração tanto a farmacodinâmica do medicamento quanto sua farmacocinética para determinar a dosagem correta e garantir o efeito adequado.
Last updated: Mar 27, 2025
A farmacocinética e a farmacodinâmica são campos de estudo que se concentram na interação entre os fármacos e o corpo.
A farmacocinética é o estudo de como o corpo humano interage com um fármaco:
A farmacodinâmica é o estudo dos efeitos de um fármaco e do seu mecanismo de ação órgão-específico, incluindo efeitos ao nível celular:
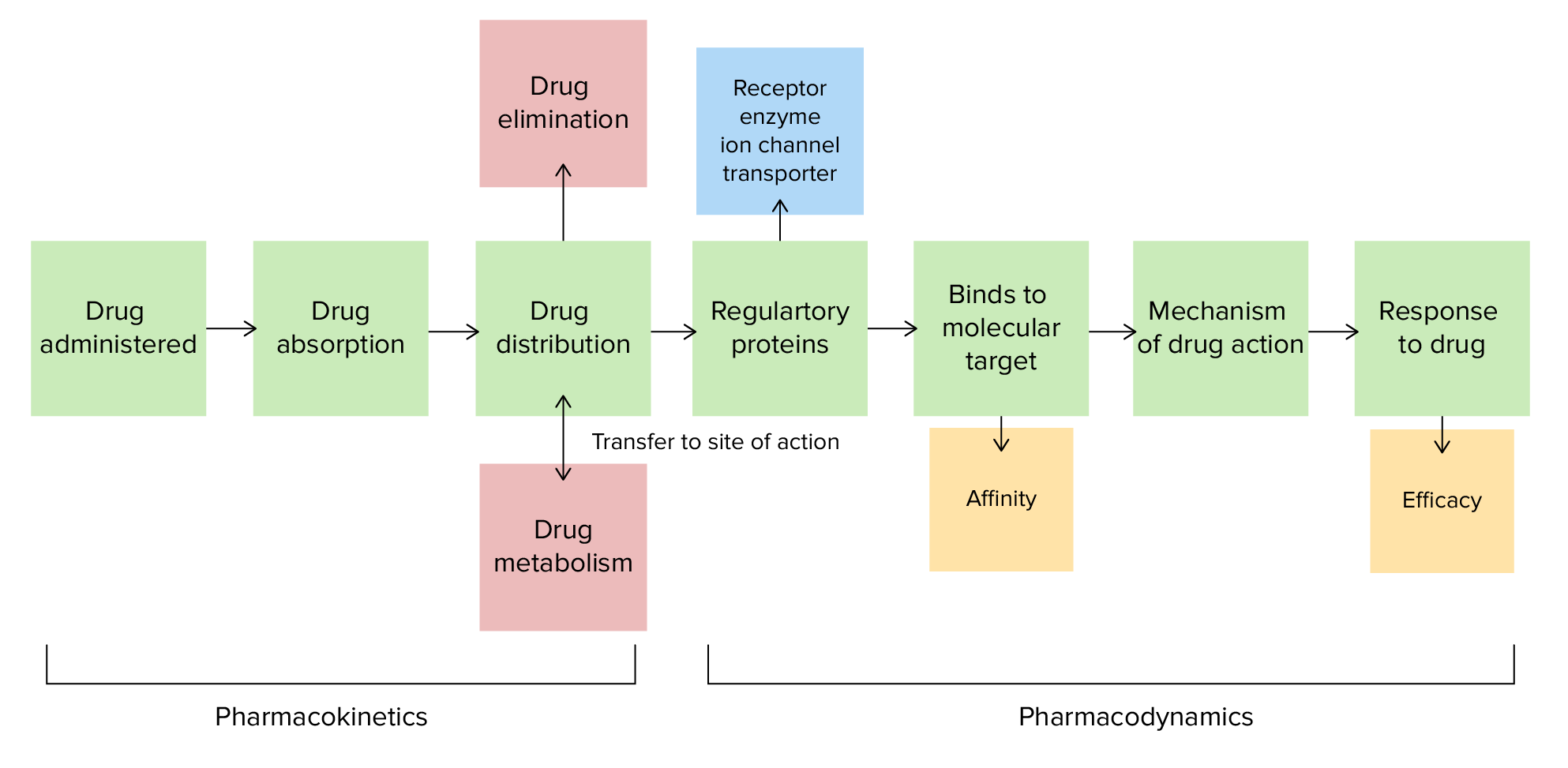
Farmacocinética e farmacodinâmica
Imagem por Lecturio.A absorção é a transferência de um fármaco ou de substância do local de administração para a corrente sanguínea e é determinada por:
A absorção através do trato GI é afetada por:
Os fármacos atravessam as membranas através de:
D: constante de difusão para o fármaco
A: área de superfície da membrana
T: espessura da membrana
C: gradiente de concentração
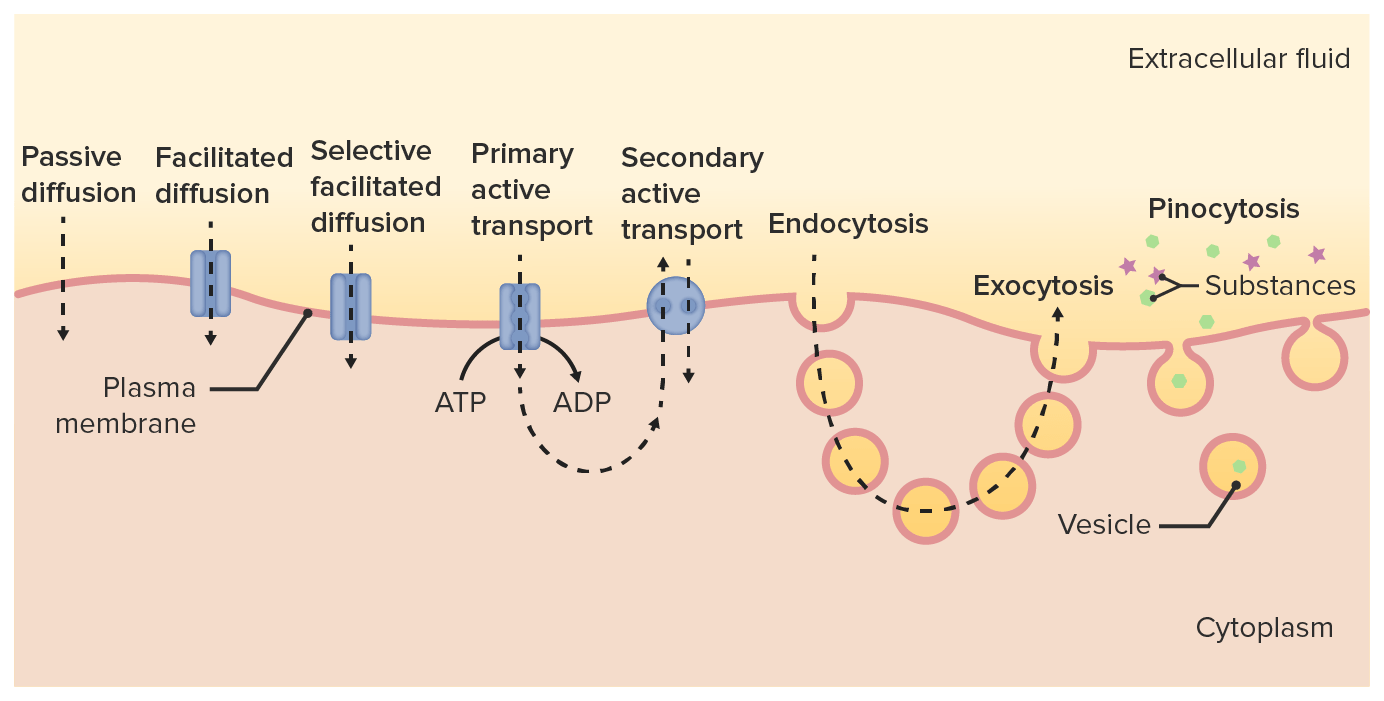
Diferentes vias de transporte de fármacos através da membrana celular e para o citoplasma
Imagem por Lecturio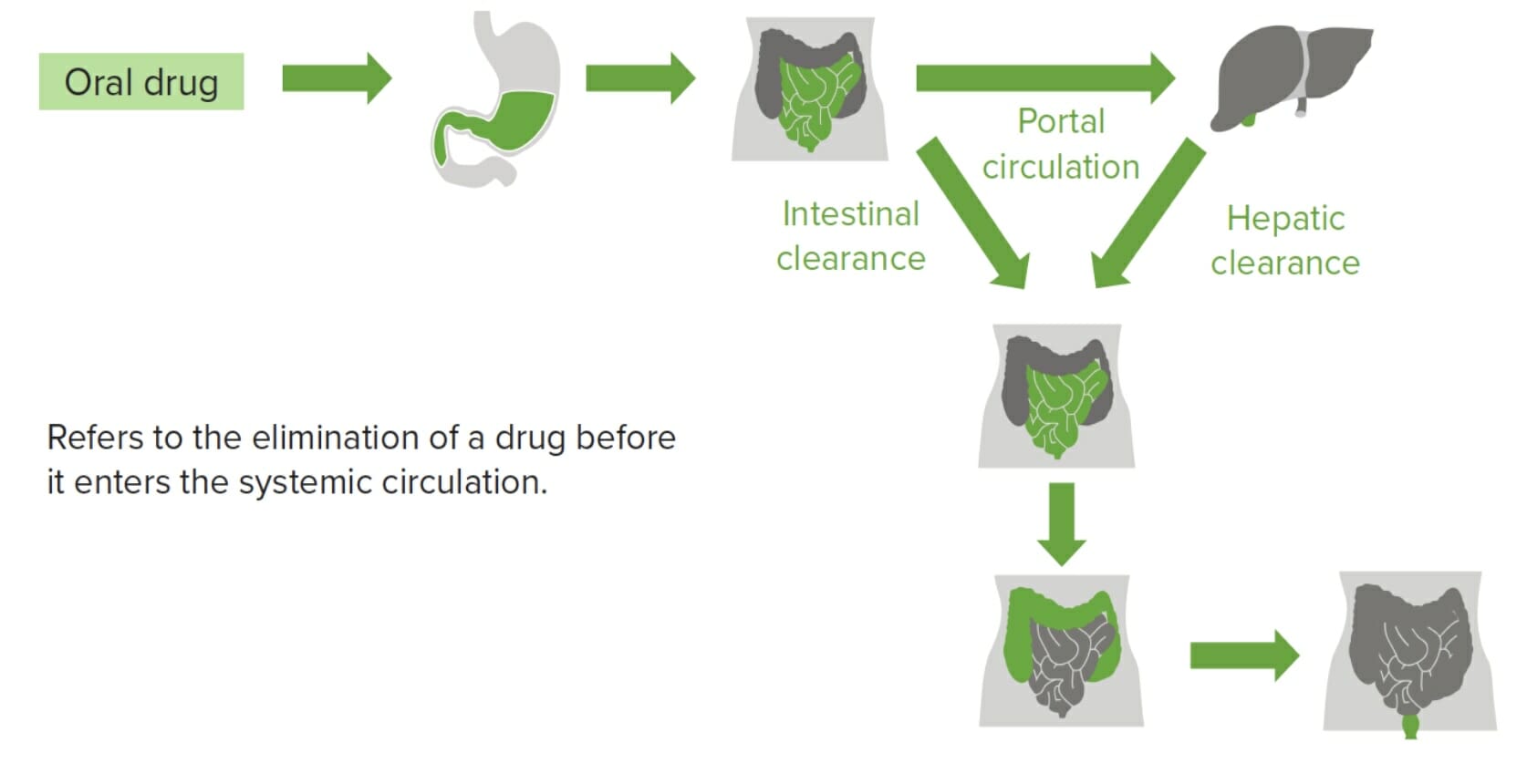
Fenómeno de um fármaco a sofrer metabolismo de primeira passagem em que é parcial ou completamente metabolizado na parede intestinal, ou é absorvido no intestino e entra na circulação portal, viajando para o fígado onde o fármaco continua a ser metabolizado.
Imagem por Lecturio.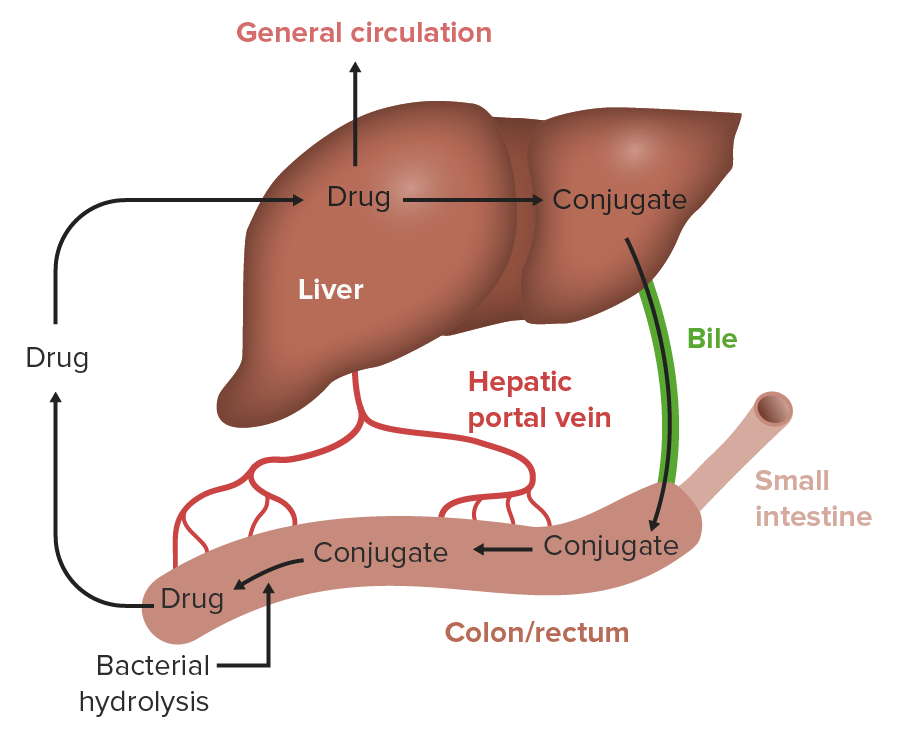
Circulação enterohepática
Imagem por LecturioA distribuição é a extensão em que um fármaco é transportado da circulação sistémica para os tecidos e órgãos-alvo.
A equação para o volume de distribuição:
$$ V_{d}= \frac{Quantidade\ do\ fármaco\ no\ organismo}{Concentração\ no\ sangue} $$A biotransformação é o processo pelo qual o corpo humano transforma quimicamente fármacos em diferentes moléculas para tornar o composto farmacologicamente ativo ou para facilitar a sua eliminação.
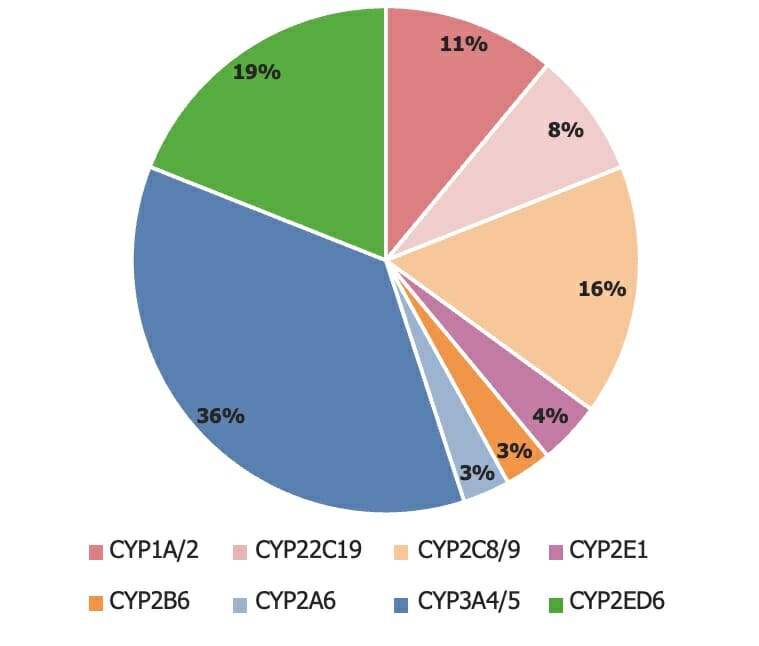
A variedade de isoenzimas do citocromo P450 (CYP450)
Imagem por Lecturio.Os recetores são macromoléculas envolvidas na sinalização química entre e dentro das células.
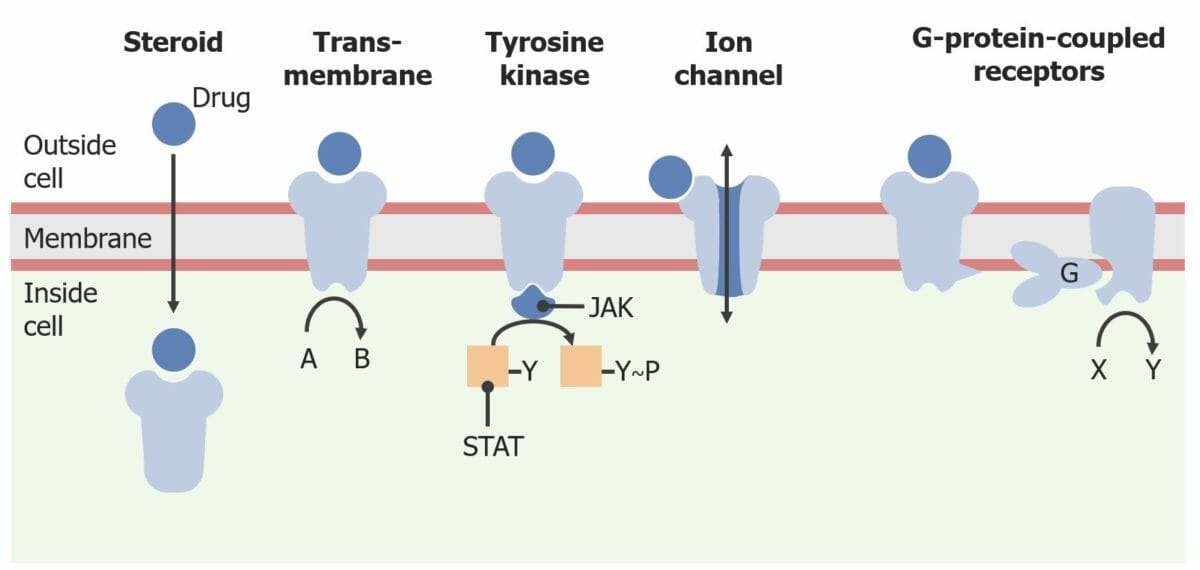
Diferentes categorias de recetores de superfície celular e intracelulares
Imagem por Lecturio.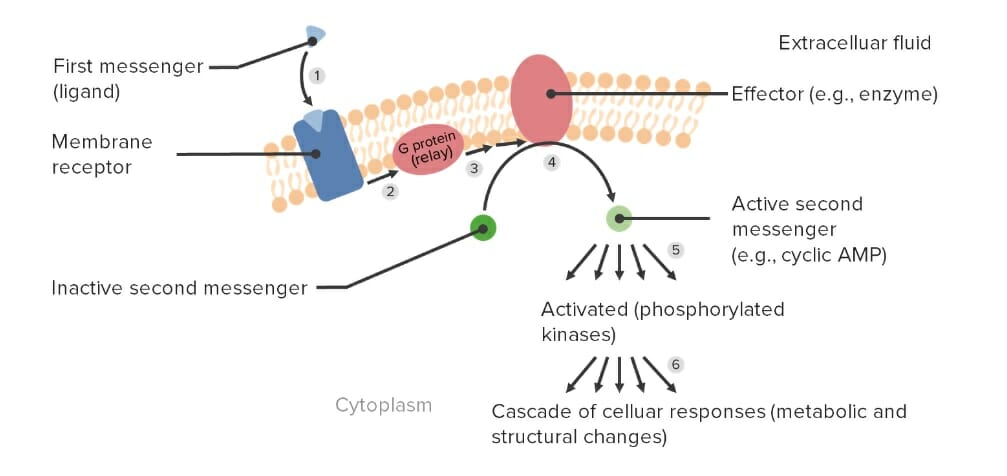
Transdução de sinal mediada por uma proteína G e ativação de um sistema de 2º mensageiro:
(1) O primeiro mensageiro ou fármaco liga-se ao recetor acoplado à proteína G e (2) ativa a proteína G, que (3) retransmite um sinal para (4) ativar a molécula efetora.
O efetor então (5) ativa 2º mensageiros para (6) eliciar respostas celulares.
O efeito de um fármaco é a resposta física que ele provoca. Este pode ser um efeito desejado (terapêutico) ou um efeito indesejado (tóxico). O efeito pode ser modulado pela presença de antagonistas e também é determinado pela sua afinidade ao seu recetor molecular alvo. Estes efeitos são medidos e podem ser representados visualmente através de curvas.
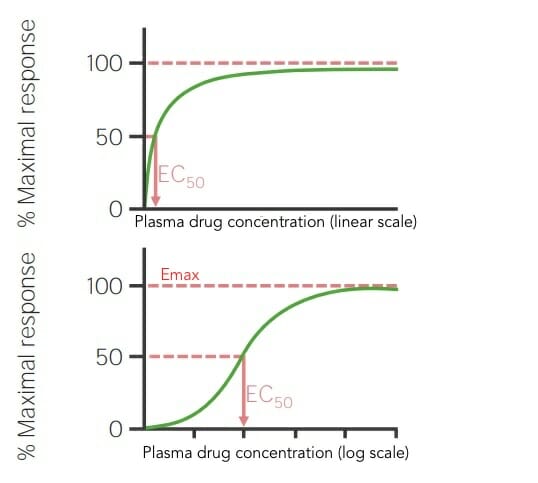
Curvas de dose-resposta em escala linear e logarítmica mostrando a dose ou concentração do fármaco que provoca um efeito máximo (Emax) e a concentração do fármaco que produz 50% do efeito máximo (EC50)
Imagem por Lecturio.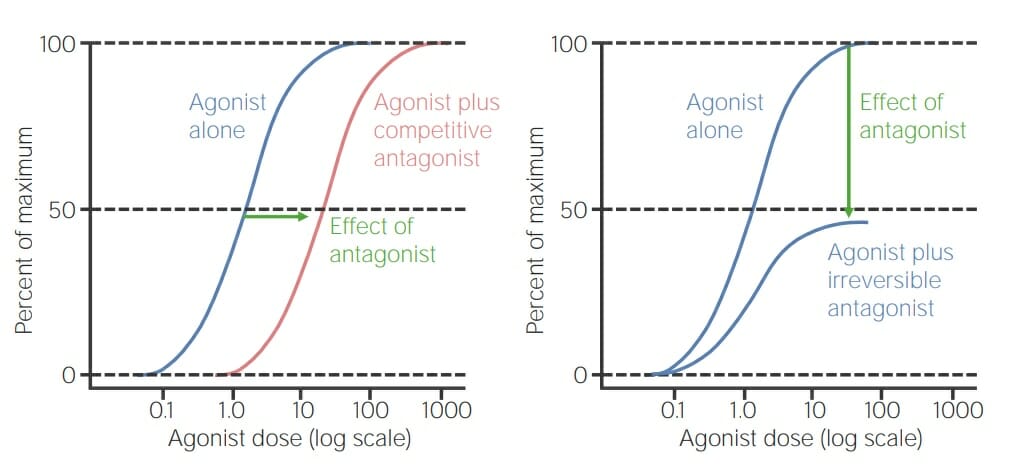
Curva dose-resposta comparando um agonista sozinho versus agonista com um antagonista competitivo (esquerda):
Observe que ocorre o mesmo efeito com uma dose mais alta de agonista quando o antagonista competitivo é dissociado. A curva é simplesmente deslocada para a direita.
Curva da direita: em comparação, esta é uma curva dose-resposta que compara agonista sozinho versus agonista com um antagonista irreversível. As 2 curvas começam na mesma concentração, mas atingem pontos máximos diferentes, uma vez que a ação do antagonista irreversível é independente da concentração do agonista. A resposta é reduzida para 50% do seu potencial máximo.
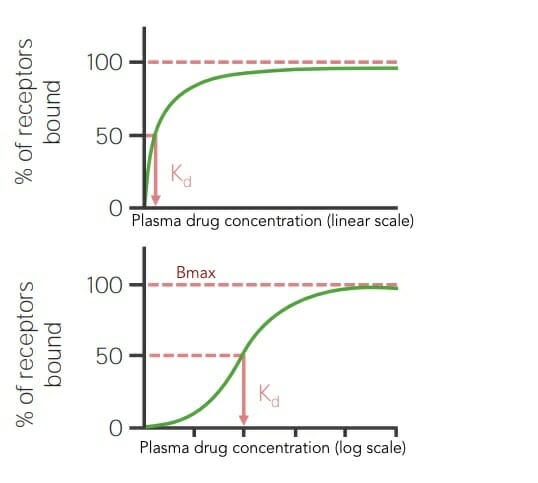
Curvas de ligação em escala linear e logarítmica mostrando a resposta biológica máxima (Bmax) e a constante de dissociação (Kd) para um fármaco
Imagem por Lecturio.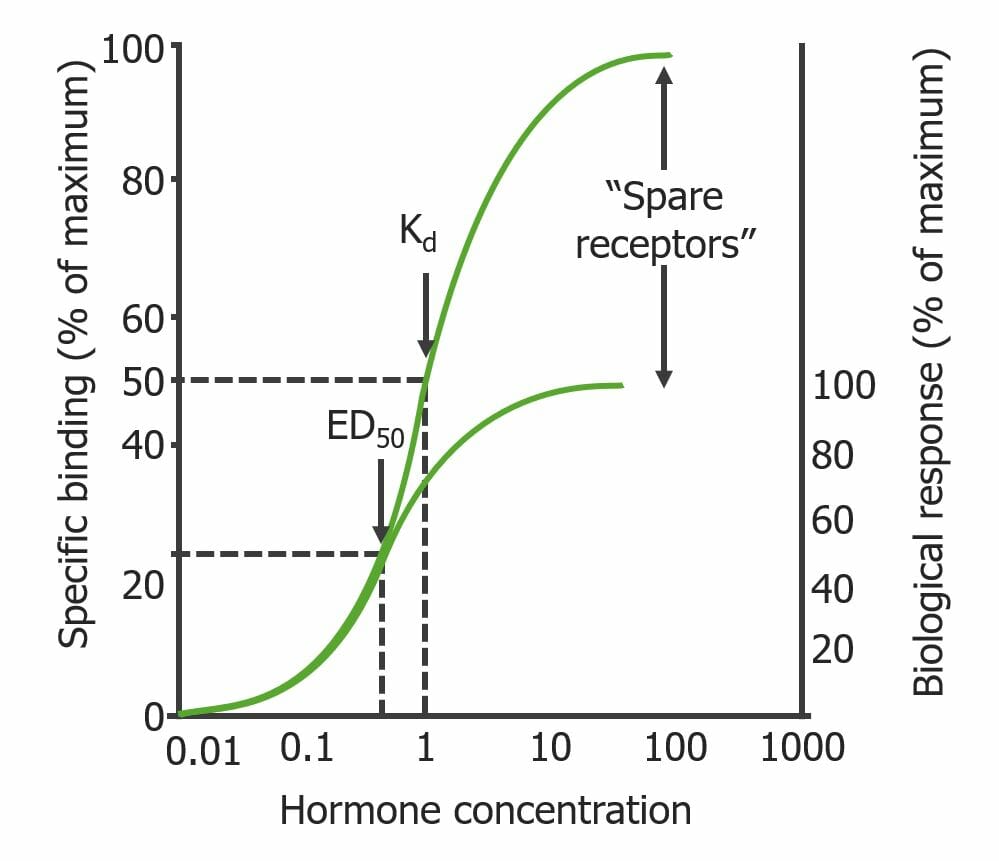
Curva de ligação que ilustra o fenómeno do recetor “de reserva”:
Há uma resposta biológica máxima apesar da presença de recetores que não estão ligados ao fármaco. Observe que a concentração do fármaco necessária para ocupar 50% dos recetores (Kd) é maior do que a concentração necessária para induzir uma resposta máxima (ED50): Kd > ED50.
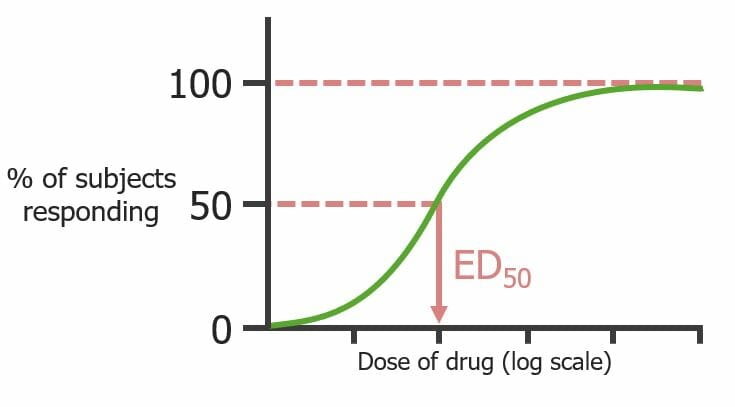
Curva dose-resposta quântica (observa a população, não os recetores individuais) evidenciando a dose de um fármaco que produz um efeito predeterminado em 50% dos indivíduos (E50)
Imagem por Lecturio.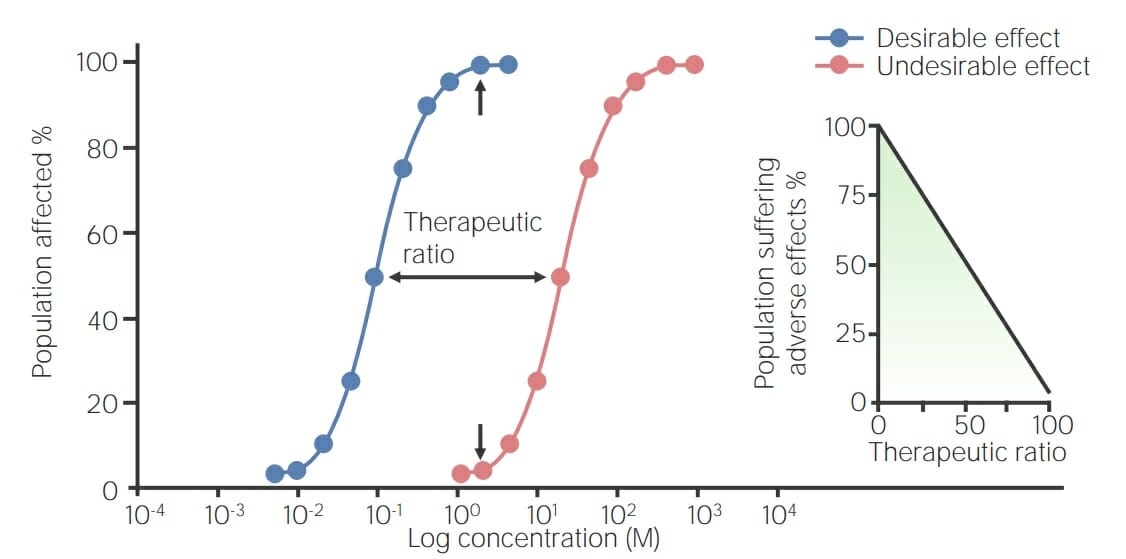
Gráfico de uma curva de toxicidade:
A curva de dose-resposta azul representa o efeito desejado de um fármaco numa população, e a curva de dose-toxicidade vermelha representa o efeito indesejável do fármaco. A razão terapêutica, ou índice terapêutico (TI, pela sigla em inglês), situa-se entre as 2 curvas e é igual à dose para um efeito tóxico em 50% da população/concentração de fármaco que produz 50% do efeito máximo (TD50 /EC50), começando nos 50% de dose efetiva máxima e terminando na dose tóxica de 50%. A inserção mostra a relação entre a proporção terapêutica e os efeitos adversos observados. Quanto maior a razão terapêutica, menor a ocorrência de efeitos adversos e vice-versa.
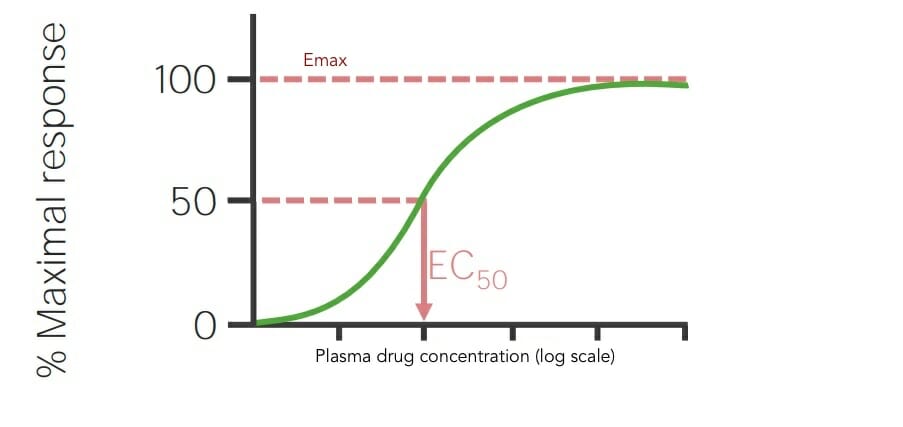
Ilustração das curvas de dose-resposta de diferentes fármacos para comparação da sua concentração necessária para produzir um efeito máximo de 50% (EC50):
Emax é o efeito máximo. Menor EC50 = maior potência. O fármaco mais à esquerda no gráfico (representada pela linha pontilhada cinzenta) tem a maior potência dos 4 fármacos exibidos porque tem a concentração mais baixa (indicada no eixo x) necessária para produzir um efeito máximo de 50%. Deslocando-se das curvas da esquerda para a direita, a potência dos fármacos diminui, com a linha cinza sólida no extremo direito sendo o fármaco menos potente.
A eliminação é o processo de conversão de um fármaco em metabolitos inativos, que são excretados do organismo no fim do processo.
Taxa de eliminação do fármaco (massa/tempo) = depuração x concentração.
Geral:
Cálculo da depuração renal:
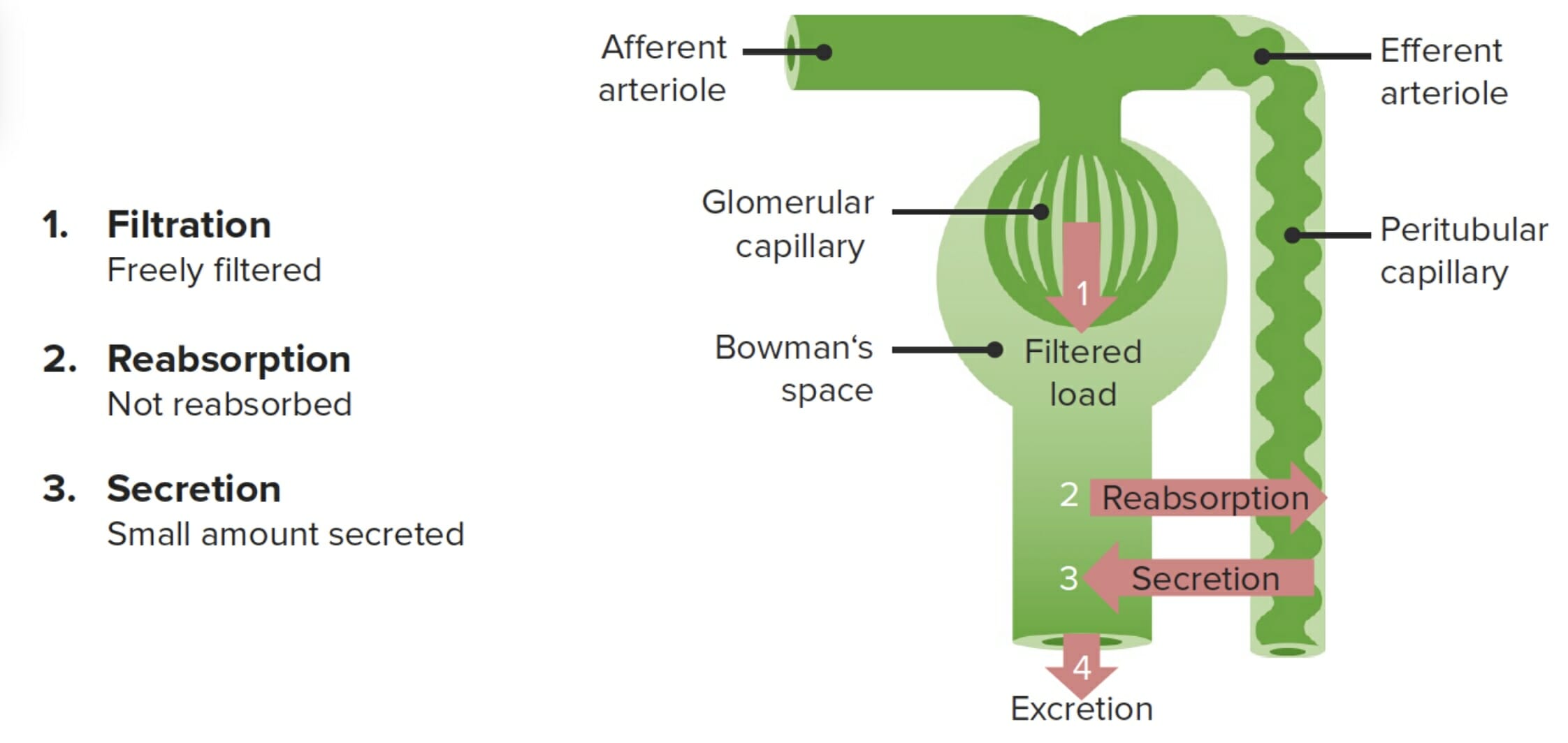
A creatinina é o marcador de filtração renal primário usado clinicamente para aproximar a taxa de filtração glomerular (TFG):
A creatinina é filtrada livremente e não é reabsorvida. No entanto, a creatinina também é secretada pelos capilares peritubulares, causando uma sobrestimação de cerca de 10% da TFG.
| Horas | Quantidade de fármco (mg/L) restante no corpo | % de fármaco eliminada | Quantidade de fármaco (mg/L) eliminada |
|---|---|---|---|
| 0 | 1 | – | – |
| 1 | 0,85 | 15 | 0,15 |
| 2 | 0,70 | 18 | 0,15 |
| 3 | 0,55 | 21 | 0,15 |
| 4 | 0,40 | 27 | 0,15 |
| 5 | 0,25 | 38 | 0,15 |
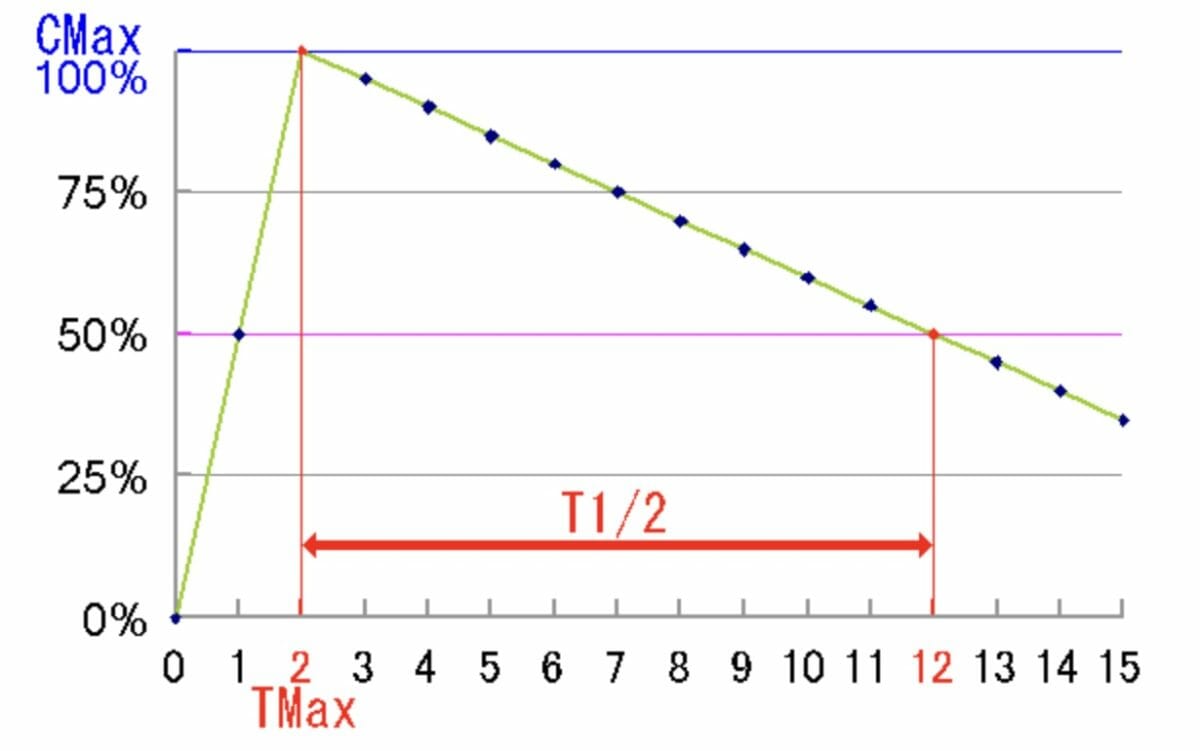
Representação gráfica da semi-vida de um fármaco:
Neste exemplo, a semi-vida é de 10 horas. Cmax = concentração máxima do fármaco na corrente sanguínea, Tmax = tempo em que a concentração do fármaco no sangue é máxima. T1/2 = semi-vida, o tempo que demora para o nível do fármaco ir de Cmax a ½ de Cmax .

Representação gráfica do fármaco na tabela acima que sofre cinética de eliminação de ordem zero:
O eixo x é a unidade de tempo em horas e o eixo y é a quantidade de fármaco, em miligramas, que permanece no organismo (ou concentração plasmática do fármaco). No tempo zero, há 1,0 mg de fármaco presente e é eliminada uma quantidade igual de fármaco a cada hora – neste exemplo, 0,15 mg.
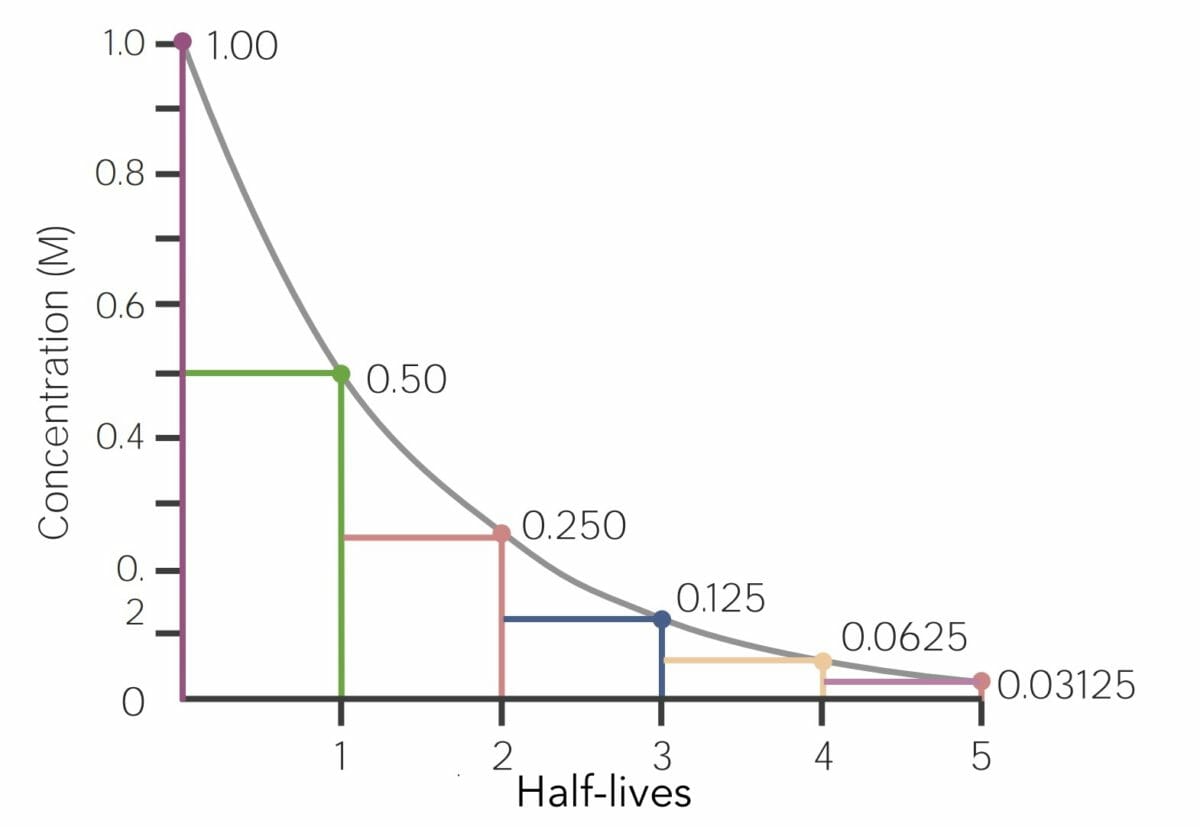
Exemplo de representação gráfica de um fármaco que sofre cinética de eliminação de 1ª ordem:
O eixo x é a unidade de tempo em horas e o eixo y é a quantidade de fármaco, em miligramas, que permanece no organismo (ou concentração plasmática de fármaco). No tempo zero, há 1,0 mg de fármaco presente, e então a cada hora é eliminada uma percentagem/proporção igual do fármaco (neste exemplo, 50%).
