La farmacocinética es la ciencia que analiza cómo el cuerpo humano interactúa con un medicamento. La farmacocinética examina cómo el cuerpo absorbe, distribuye, metaboliza y excreta el medicamento. La farmacodinamia es la ciencia que estudia los efectos bioquímicos y fisiológicos de un medicamento y su mecanismo de acción específico de un órgano, incluidos los efectos a nivel celular. Otra forma de describir la diferencia entre las 2 disciplinas es decir que la farmacocinética es "lo que el cuerpo le hace al medicamento", mientras que la farmacodinamia es "lo que el medicamento le hace al cuerpo". AlALAmyloidosis prescribir medicamentos, losLOSNeisseria médicos deben tener enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum cuenta tanto la farmacodinamia como la farmacocinética del medicamento para determinar la dosis correcta y garantizar el efecto adecuado.
La farmacocinética y la farmacodinamia son campos de estudio que se centran enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la interacción entre losLOSNeisseria medicamentos y el cuerpo.
La farmacocinética es el estudio de cómo el cuerpo humano interactúa con un medicamento:
Absorción
Distribución
Metabolismo/eliminación
La farmacodinamia es el estudio de losLOSNeisseria efectos de un medicamento y su mecanismo de acción específico enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum cada órgano, incluidos losLOSNeisseria efectos a nivel celular:
Dinámica de unión medicamento-receptor
Mecanismo de acción del medicamento
Respuesta fisiológica
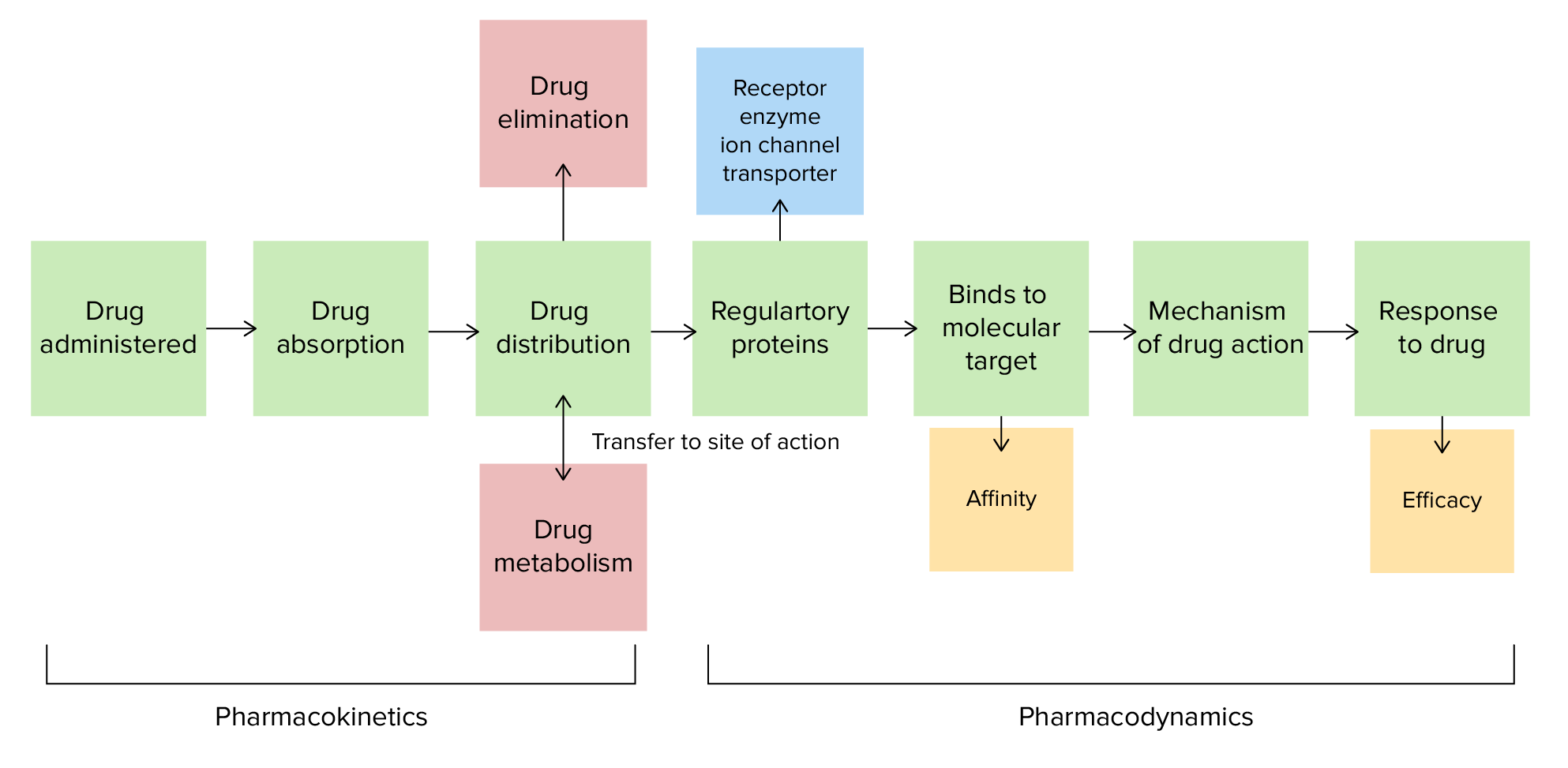
Farmacocinética y farmacodinamia
Imagen por Lecturio.
Farmacocinética: Absorción
Definición
La absorción es la transferencia de un medicamento o sustancia desde el sitio de administración alALAmyloidosis torrente sanguíneo y está determinada por:
Las propiedades fisicoquímicas del medicamento:
Solubilidad enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum lípidos
Diferencias enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance luminal a lo largo del tracto gastrointestinal:
La mayoría de losLOSNeisseria medicamento son ácidos o basesBasesUsually a hydroxide of lithium, sodium, potassium, rubidium or cesium, but also the carbonates of these metals, ammonia, and the amines.Acid-Base Balance orgánicas débiles.
LosLOSNeisseria medicamento existen enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum formas no ionizadas y ionizadas.
La forma no ionizada es generalmente soluble enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum lípidos (lipofílica) y se difunde fácilmente a través de las membranas celulares.
La forma ionizada tiene alta solubilidad enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum agua (hidrofílica).
La proporción de la forma no ionizada está determinada por:
El pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance ambiental
El pKa (constante de disociación ácida) del medicamento: el pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base BalanceenENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el que las concentraciones de formas ionizadas y no ionizadas son iguales
Superficie por volumen luminal:
El intestino delgado tiene la mayor superficie.
La mayor parte de la absorción se produce enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el intestino delgado.
Perfusión sanguínea de la membrana absorbente:
La disminución del flujo sanguíneo (e.g., enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el shockShockShock is a life-threatening condition associated with impaired circulation that results in tissue hypoxia. The different types of shock are based on the underlying cause: distributive (↑ cardiac output (CO), ↓ systemic vascular resistance (SVR)), cardiogenic (↓ CO, ↑ SVR), hypovolemic (↓ CO, ↑ SVR), obstructive (↓ CO), and mixed. Types of Shock) reduce la absorción.
Presencia de bilis y moco:
EnENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el estómago, una gruesa capa de moco limita la absorción del medicamento.
Reacciones químicas:
Hidrólisis por ácido gástrico o enzimas digestivas
Metabolismo por la flora bacteriana del tracto gastrointestinal
Tiempo de tránsito:
El tránsito rápido (e.g., estados diarreicos) a través del tracto gastrointestinal dificultará la absorción.
El retraso enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el vaciamiento gástrico afectará la absorción por el intestino delgado.
La naturaleza de las membranas epiteliales (véase más abajo)
LosLOSNeisseria medicamento atravesarán las membranas a través de:
Difusión pasiva, dependiendo de:
Grado de gradiente de concentración a través de la membrana: losLOSNeisseria medicamento se mueven de áreas de alta a baja concentración.
Liposolubilidad del medicamento: debido a que las membranas son lipídicas, losLOSNeisseria medicamento solubles enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum lípidos se difunden más rápidamente.
Tamaño de las partículas del medicamento: las partículas pequeñas penetran las membranas más rápidamente.
Grado de ionización: porque determina la liposolubilidad
Área de superficie de absorción: cuanto mayor es la superficie, mayor es la difusión
La ley de Fick gobierna la difusión pasiva, mostrando que la tasa de difusión es proporcional a:
$$ V_{líquido}= D \frac{A}{T}(C_{1}-C_{2}) $$
D: constante de difusión del medicamento A: área de superficie de la membrana T: espesor de la membrana C: gradiente de concentración
Difusión pasiva facilitada:
Requiere la presencia de una molécula portadora que se una alALAmyloidosis medicamento y lo transporte a través de la membrana
La difusión todavía ocurre a lo largo de un gradiente de concentración.
No requiere gasto energético
Transporte activo:
Requiere gasto energético
Puede ocurrir contra un gradiente de concentración
Ocurre con medicamentos que son similares a sustancias endógenas—i.e., vitaminas, azúcares, aminoácidos
Pinocitosis:
El líquido o las partículas son endocitados por la célula enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum forma de vesículas
Requiere gasto energético
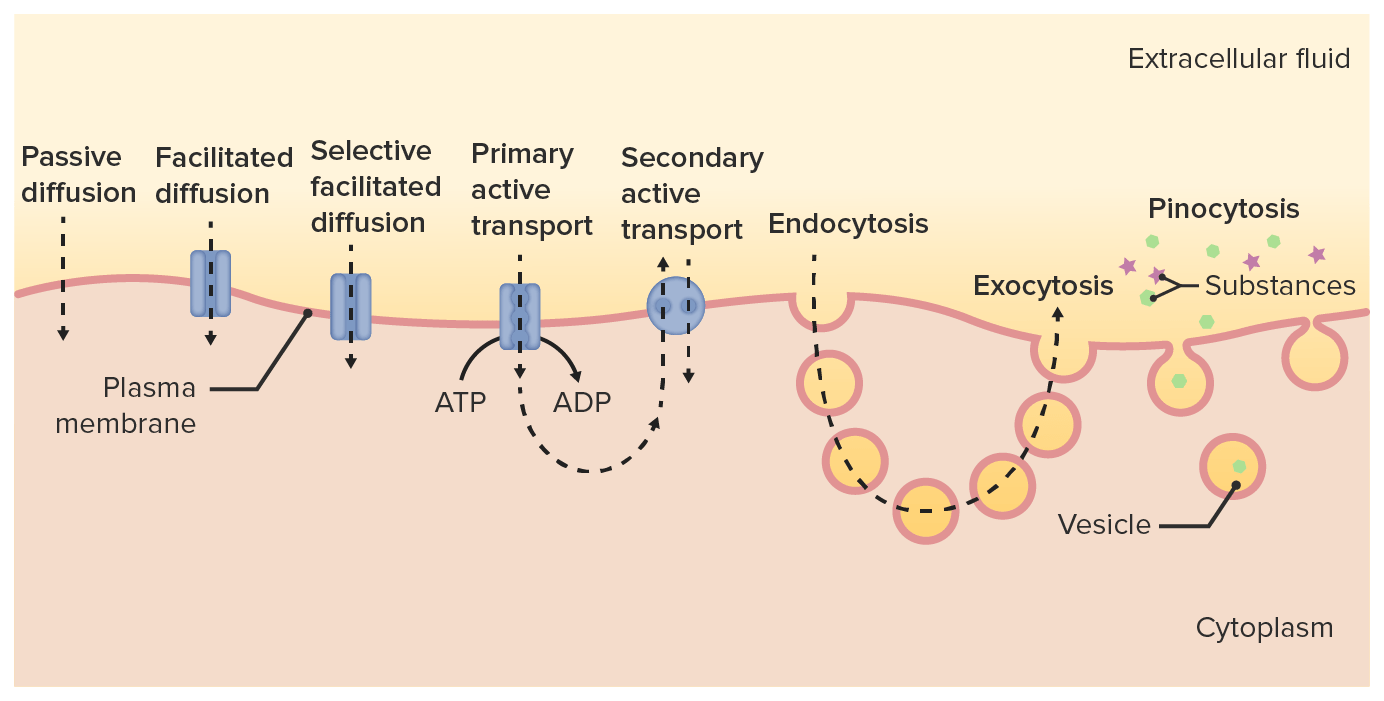
Diferentes vías de transporte de fármacos a través de la membrana celular y hacia el citoplasma
Imagen por Lecturio
Biodisponibilidad
La extensión y la velocidad a la que un medicamento entra enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la circulación sistémica, accediendo así alALAmyloidosis sitio de acción
Relevante solo para medicamentos administrados por vía oral, ya que losLOSNeisseria medicamentos intravenosos tienen una biodisponibilidad del 100%
Factores que afectan la biodisponibilidad:
Cualquier cosa que afecte la absorción
Metabolismo hepático de 1er paso
LosLOSNeisseria medicamento absorbidos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el tracto gastrointestinal pasan por la circulación portal y terminan enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el hígado.
El hígado metaboliza el medicamento antes de que entre enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la circulación sistémica, lo que reduce la biodisponibilidad del medicamento.
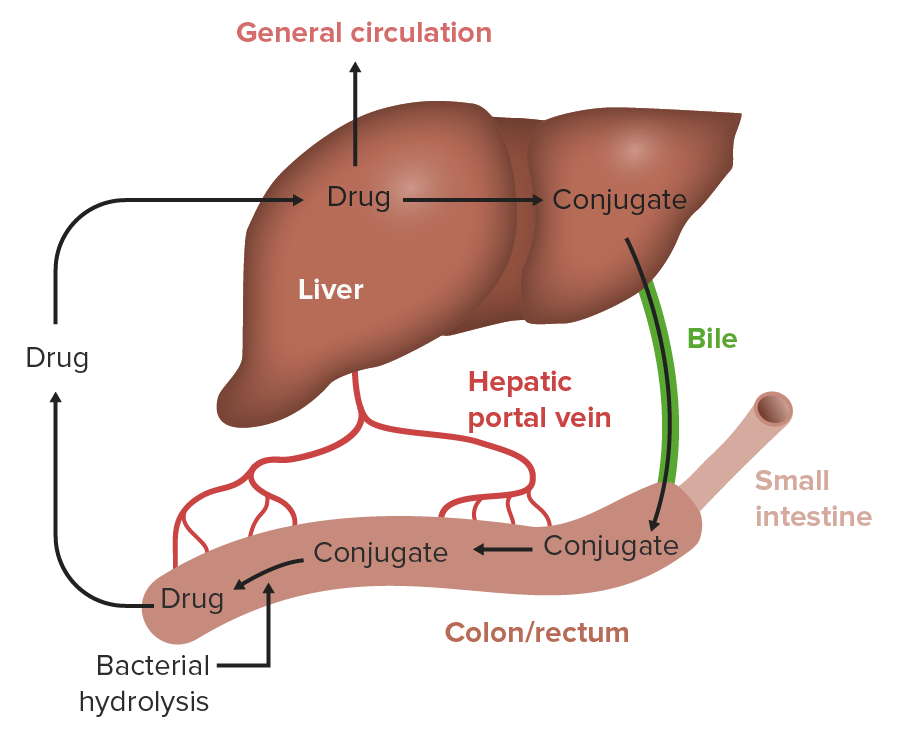
Circulación enterohepática:
El medicamento se absorbe enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el tracto gastrointestinal y, a través de la circulación portal, es captado por el hígado.
El medicamento activo o sus metabolitos se excretan enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la bilis y luego enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el intestino.
La microbiota intestinal desconjuga losLOSNeisseria metabolitos de losLOSNeisseria medicamentos para liberar la molécula original del medicamento.
Luego, el medicamento se reabsorbe enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el intestino → se reinicia el ciclo
La recirculación puede producir múltiples picos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la concentración plasmática del medicamento.
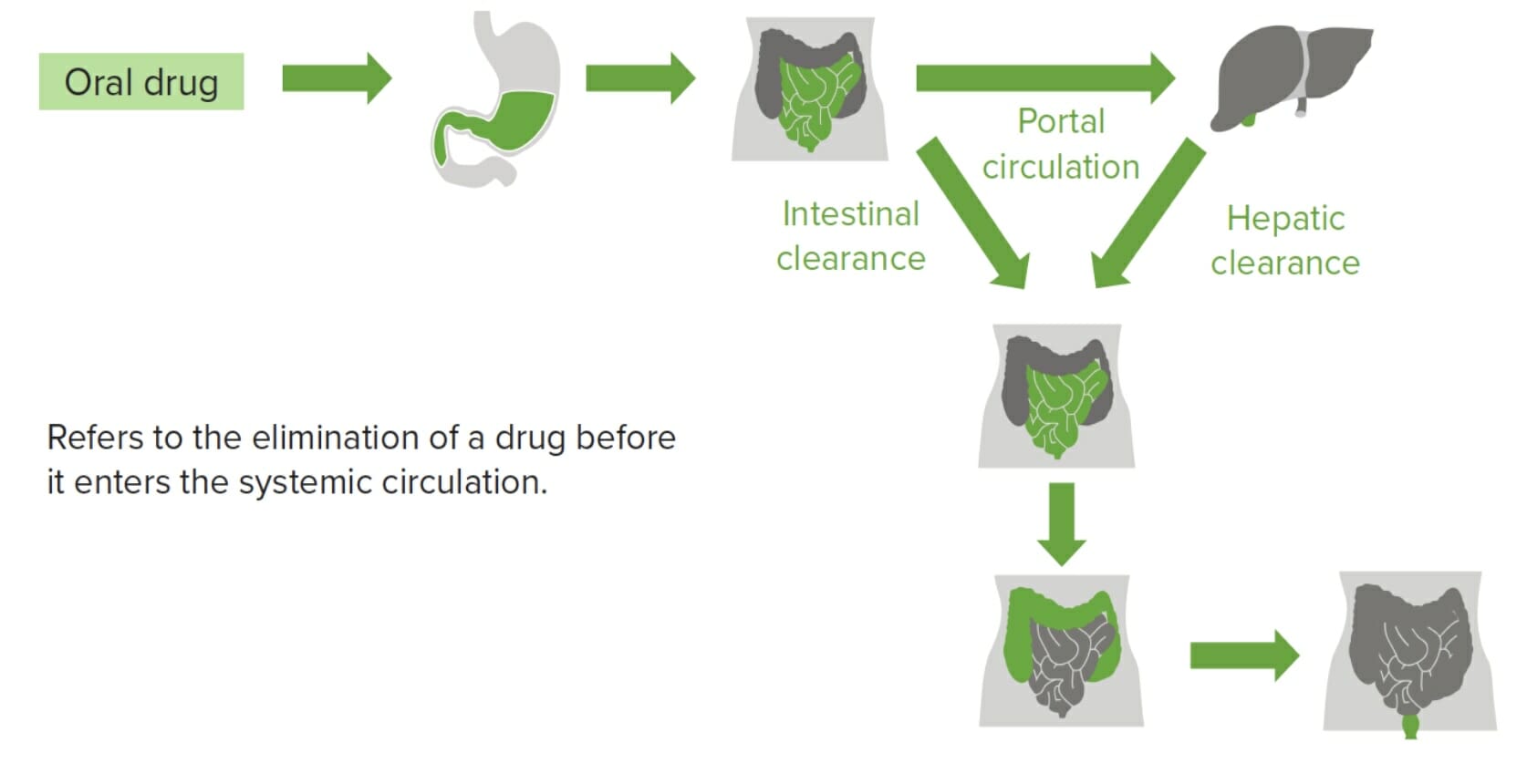
Fenómeno en el que un fármaco se somete al metabolismo de primer paso en el que se metaboliza parcial o totalmente en la pared intestinal, o se absorbe en el intestino y entra en la circulación portal y viaja al hígado donde el fármaco se metaboliza aún más.
La distribución esla medida enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum que un medicamento se transporta desde la circulación sistémica a losLOSNeisseria tejidos y órganos diana.
Volumen de distribución (Vd)
Volumen necesario para contener la cantidad total de un medicamento a la misma concentración enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum que se observa enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products
También se puede conceptualizar como la relación entre la cantidad de medicamento enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum un cuerpo (dosis) y la concentración del medicamento medida enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum sangre y plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products y libre enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el líquido intersticial
Un volumen teórico que proporciona una referencia para la concentración plasmática esperada para una dosis dada
Factores que afectan la distribución de medicamentos
Permeabilidad tisular de medicamento; depende de las características del medicamento:
Tamaño molecular: las moléculas más pequeñas se distribuyen enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum mayor medida enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria tejidos.
pKa: determina el grado de ionización y, por lo tanto, la lipofilia:
LosLOSNeisseria medicamentos lipofílicos se disuelven enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum lípidos: difusión de lípidos
LosLOSNeisseria medicamentos lipofílicos pueden atravesar las membranas celulares lipídicas.
LosLOSNeisseria medicamentos lipofílicos pueden atravesar las barreras hematoencefálica y placentaria.
LosLOSNeisseria medicamentos hidrofílicos se disuelven enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum agua: difusión acuosa
LosLOSNeisseria medicamentos hidrofílicos requieren poros o transportadores para atravesar las membranas.
Barreras tisulares:
Barrera hematoencefálica: enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria capilares cerebrales, las células endoteliales están conectadas por uniones intercelulares estrechas.
Barrera placentaria: formada por la membrana basal trofoblástica fetal y una capa endotelial
Barrera hematotesticular: formada por uniones estrechas entre las células testiculares de Sertoli
Gasto cardíaco/flujo sanguíneo regional:
Cuanto mayor sea el flujo sanguíneo del órgano, mayor será la distribución del medicamento
Cuanto más grande es el órgano, mayor es el flujo de sangre hacia el mismo
Grado de unión a proteínas:
La albúmina es la principal proteína de unión a medicamentos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products.
Otras proteínas de unión son la glicoproteína ácida alfa-1 y las lipoproteínas.
Solo el medicamento libre puede difundirse pasivamente a sitios extravasculares o tisulares para ejercer sus efectos.
La concentración de medicamento libre enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products determina la concentración del medicamento enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el sitio activo y la eficacia.
LosLOSNeisseria medicamentos unidos a la albúmina permanecen intravasculares.
La alta unión a proteínas disminuye el volumen de distribución, ya que losLOSNeisseria medicamentos no pueden difundirse enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria tejidos.
LosLOSNeisseria medicamentos libres pueden difundirse y unirse a losLOSNeisseria tejidos y tener un gran volumen de distribución.
Composición corporal:
Agua extracelular: afecta el volumen de distribución de medicamentos hidrofílicos
Tejido adiposo: afecta el volumen de distribución de medicamentos lipofílicos
Edad:
El agua corporal total es más alta enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria lactantes.
El contenido de grasa es mayor enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum lactantes y adultos mayores.
El músculo esquelético es menor enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum lactantes y adultos mayores.
Composición de órganos: sistema nervioso inmaduro enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum lactantes → mayor distribución de medicamentos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el cerebro
El contenido de proteína plasmática es menor enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum lactantes y adultos mayores.
Sexo:
Las mujeres tienen menos agua corporal total, debido a su tamaño más pequeño, y más tejido adiposo.
Embarazo:
El aumento del volumen de sangre conduce a un mayor Vd.
El feto es un compartimento separado enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el que se pueden distribuir losLOSNeisseria medicamentos.
Obesidad:
El alto tejido adiposo conduce a una mayor distribución y acumulación de medicamentos lipofílicos.
LosLOSNeisseria estados patológicos pueden afectar:
Concentraciones de albúmina plasmática
Perfusión tisular: i.e., hipoperfusión enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el shockShockShock is a life-threatening condition associated with impaired circulation that results in tissue hypoxia. The different types of shock are based on the underlying cause: distributive (↑ cardiac output (CO), ↓ systemic vascular resistance (SVR)), cardiogenic (↓ CO, ↑ SVR), hypovolemic (↓ CO, ↑ SVR), obstructive (↓ CO), and mixed. Types of Shock
pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance tisular: i.e., acidosisAcidosisA pathologic condition of acid accumulation or depletion of base in the body. The two main types are respiratory acidosis and metabolic acidosis, due to metabolic acid build up.Respiratory Acidosis láctica por sepsisSepsisSystemic inflammatory response syndrome with a proven or suspected infectious etiology. When sepsis is associated with organ dysfunction distant from the site of infection, it is called severe sepsis. When sepsis is accompanied by hypotension despite adequate fluid infusion, it is called septic shock.Sepsis and Septic Shock
Alteración de las barreras fisiológicas: la meningitisMeningitisMeningitis is inflammation of the meninges, the protective membranes of the brain, and spinal cord. The causes of meningitis are varied, with the most common being bacterial or viral infection. The classic presentation of meningitis is a triad of fever, altered mental status, and nuchal rigidity. Meningitis altera la barrera hematoencefálica.
Dieta:
Una dieta rica enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum grasas aumenta losLOSNeisseria ácidos grasos libres, que compiten con losLOSNeisseria medicamentos para unirse a la albúmina.
La desnutrición disminuye losLOSNeisseria niveles de albúmina enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumplasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products, lo que afecta la unión del medicamento y la distribución tisular.
Interacciones medicamentosas:
El desplazamiento se produce cuando se administran de forma concomitante 2 medicamentos que tienen la misma afinidad por el mismo sitio de unión.
La biotransformación esel proceso a través del cual el cuerpo humano transforma químicamente losLOSNeisseria medicamentos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum diferentes moléculas para hacer que el compuesto sea farmacológicamente activo o para facilitar la eliminación.
El metabolismo es un tipo de biotransformación.
Generalmente, tiene lugar enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el hígado, a través de enzimas hepáticas
Cuando un medicamento o compuesto original se metaboliza, se puede convertir enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum:
Forma inactiva
Forma farmacológicamente activa
Metabolito tóxico
Fases de la biotransformación
Reacciones de fase I: transforma el medicamento enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum un metabolito polar, lo que lo haceHACEAltitude Sickness más soluble enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum agua
Esto se haceHACEAltitude Sickness desenmascarando o insertando un grupo polar (–OH, –SH, –NH2).
Realizado por isoenzimas del citocromo P450 (CYP450) enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el hígado
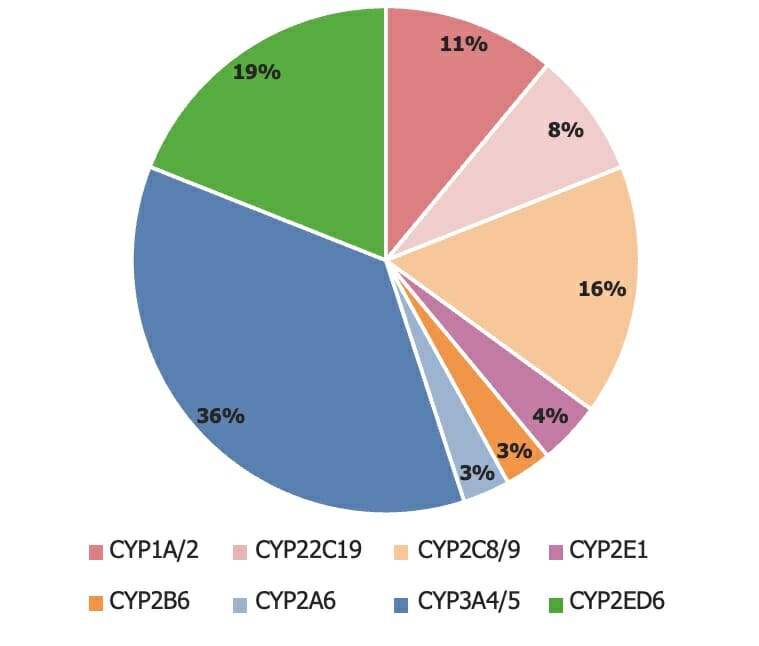
El 75% de losLOSNeisseria medicamentos son metabolizados por CYP450-3A4, CYP450-3A5 y CYP450-2D6.
Reacciones de fase II: conjugación del metabolito con compuestos para aumentar la solubilidad enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum agua, que incluyen:
Glucuronidación
Acetilación
Conjugación de glutatión
Sulfatación
Metilación
Reacciones de fase III: mayor procesamiento de losLOSNeisseria medicamentos, que incluye:
Preparación de un medicamento para su excreción enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la bilis, la orina u otros lúmenes
Unión a proteínas de transporte, generalmente glicoproteínas P
No todos losLOSNeisseria medicamentos pasan por las 3 fases.
Existe una gran variabilidad genética enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la actividad de las enzimas involucradas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum las 3 fases.
Como resultado, losLOSNeisseria individuos pueden presentar diferencias significativas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum su capacidad para metabolizar el mismo medicamento.
Un gran número de medicamentos pueden inducir e inhibir el sistema enzimático P450.
La variedad de isoenzimas del citocromo P450 (CYP450)
Imagen por Lecturio.
Factores que afectan el metabolismo de losLOSNeisseria medicamentos
Diferencias genéticas de CYP450
Inhibición competitiva de CYP450:
LosLOSNeisseria medicamentos pueden competir por la misma vía.
Un medicamento puede inhibir o inducir el metabolismo de otro medicamento.
Inhibición directa de CYP450:
Amiodarona, ritonavirRitonavirAn HIV protease inhibitor that works by interfering with the reproductive cycle of HIV. It also inhibits cytochrome p-450 cyp3a.Anti-HIV Drugs, cimetidina, jugo de toronja, cimetidina, fluconazol
Inhibidores suicidas: inhibición irreversible de enzimas/receptores, e.g., secobarbital
Inducción de CYP450:
Medicamentos anticonvulsivantes, etanol, hierba de San Juan, rifampicina
Inhibición de la glicoproteína P (mutación multirresistente a medicamentos (MDR1))
Mueve losLOSNeisseria medicamentos desde el interior de la célula hasta el lumen intestinal
La inhibición de MDR1 aumenta losLOSNeisseria niveles intracelulares del medicamento, lo que provoca toxicidad
LosLOSNeisseria inhibidores incluyen verapamilo y jugo de toronja.
LosLOSNeisseria medicamentos metabolizados por MDR1 incluyen ciclosporina y digoxina
A concentraciones terapéuticas, el medicamento solo ocupa una pequeña fracción de losLOSNeisseria sitios de la enzima metabolizadora.
La tasa de metabolismo aumenta con la concentración del medicamento.
La tasa de metabolismo del medicamento es una fracción constante del medicamento que queda enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el cuerpo.
El medicamento tiene una vida media específica: tiempo necesario para que la concentración plasmática se reduzca a la mitad de su valor original.
Cinética de orden cero:
La mayoría de losLOSNeisseria sitios de unión a medicamentos de las enzimas metabolizadoras están ocupados.
El metabolismo funciona a su máxima velocidad y no cambia enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum proporción a la concentración del medicamento.
EnENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum este caso, no se puede determinar una vida media específica.
A medida que aumenta la concentración del medicamento, el metabolismo cambia de una cinética de 1er orden a una de orden cero.
Mnemotecnia para inductores e inhibidores de CYP450
Inductores: “PARC GPS” (enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum inglés)
P – PhenytoinPhenytoinAn anticonvulsant that is used to treat a wide variety of seizures. The mechanism of therapeutic action is not clear, although several cellular actions have been described including effects on ion channels, active transport, and general membrane stabilization. Phenytoin has been proposed for several other therapeutic uses, but its use has been limited by its many adverse effects and interactions with other drugs.First-Generation Anticonvulsant Drugs (fenitoína)
A – Alcohol (etanol)
R – RifampinRifampinA semisynthetic antibiotic produced from streptomyces mediterranei. It has a broad antibacterial spectrum, including activity against several forms of Mycobacterium. In susceptible organisms it inhibits dna-dependent RNA polymerase activity by forming a stable complex with the enzyme. It thus suppresses the initiation of RNA synthesis. Rifampin is bactericidal, and acts on both intracellular and extracellular organisms.Epiglottitis (rifampina)
C – CarbamazepineCarbamazepineA dibenzazepine that acts as a sodium channel blocker. It is used as an anticonvulsant for the treatment of grand mal and psychomotor or focal seizures. It may also be used in the management of bipolar disorder, and has analgesic properties.First-Generation Anticonvulsant Drugs (carbamazepina)
G -Griseofulvin (griseofulvina)
P – Phenobarcital (fenobarbital)
S – SmokingSmokingWillful or deliberate act of inhaling and exhaling smoke from burning substances or agents held by hand.Interstitial Lung Diseases (tabaquismo)
S – St. John’s wort (hierba de San Juan)
Inhibidores: “PACMAN-G” (enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum inglés)
P – Protease inhibitorsProtease InhibitorsCompounds which inhibit or antagonize biosynthesis or actions of proteases (endopeptidases).Anti-HIV Drugs (inhibidores de la proteasa)
A – Azole antifungals (antifúngicos azólicos)
C – CimetidineCimetidineA histamine congener, it competitively inhibits histamine binding to histamine h2 receptors. Cimetidine has a range of pharmacological actions. It inhibits gastric acid secretion, as well as pepsin and gastrin output.Antihistamines (cimetidina)
M – MacrolidesMacrolidesMacrolides and ketolides are antibiotics that inhibit bacterial protein synthesis by binding to the 50S ribosomal subunit and blocking transpeptidation. These antibiotics have a broad spectrum of antimicrobial activity but are best known for their coverage of atypical microorganisms. Macrolides and Ketolides (macrólidos)
A – AmiodaroneAmiodaroneAn antianginal and class III antiarrhythmic drug. It increases the duration of ventricular and atrial muscle action by inhibiting potassium channels and voltage-gated sodium channels. There is a resulting decrease in heart rate and in vascular resistance.Pulmonary Fibrosis (amiodarona)
N – Nondihydropyridine calciumCalciumA basic element found in nearly all tissues. It is a member of the alkaline earth family of metals with the atomic symbol ca, atomic number 20, and atomic weight 40. Calcium is the most abundant mineral in the body and combines with phosphorus to form calcium phosphate in the bones and teeth. It is essential for the normal functioning of nerves and muscles and plays a role in blood coagulation (as factor IV) and in many enzymatic processes.Electrolytes channel blockers (bloqueadores de losLOSNeisseria canales de calcio (BCC) no dihidropiridínicos)
Farmacodinamia: Receptores de Medicamentos y Efectores
LosLOSNeisseria receptores son macromoléculas involucradas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la señalización química entre y dentro de las células.
Estos receptores pueden estar ubicados enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la membrana de la superficie celular o dentro del citoplasma:
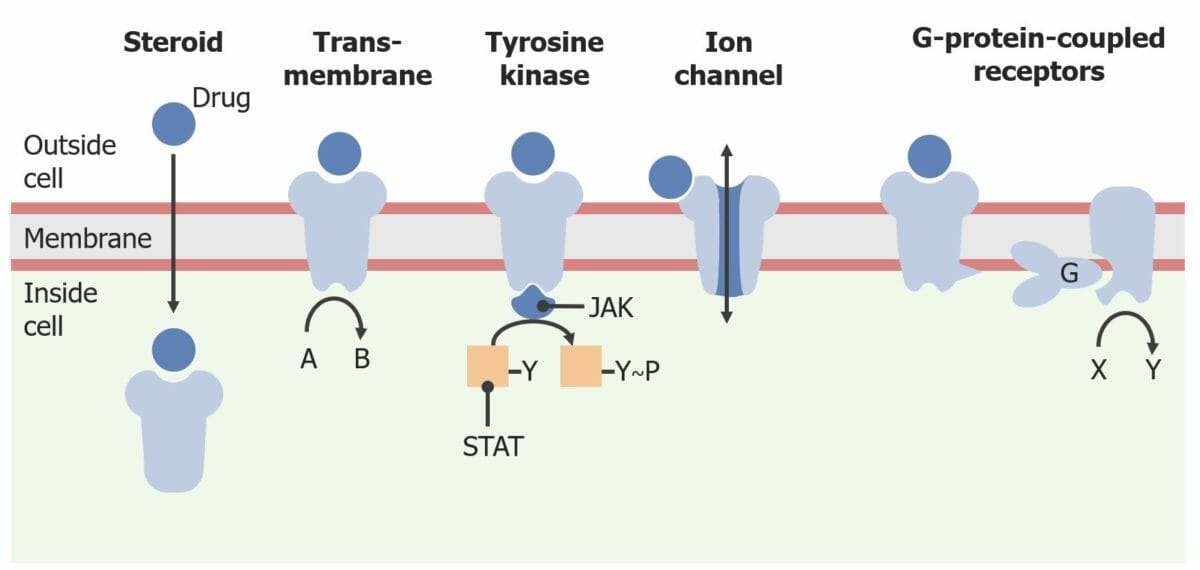
LosLOSNeisseria receptores de la superficie celular tienen una fracción transmembrana que losLOSNeisseria conecta con el citoplasma:
Receptores acoplados a proteína G
Receptores de tirosina quinasa
Canales iónicos
Receptores intracelulares (e.g., receptores de esteroides): el medicamento o ligando debe ser lipofílico.
Receptores de esteroides
Receptores de hormonas tiroideas
Receptores de vitamina A y D
Un medicamento puede unirse a una región molecular específica del receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors llamada sitio de reconocimiento.
El sitio de reconocimiento de un medicamento puede ser diferente alALAmyloidosis de otro medicamento que se une alALAmyloidosis mismo receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors.
Un medicamento que se une a un receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors puede activarlo o desactivarlo, lo que aumenta o disminuye la función:
LosLOSNeisseria agonistas activan losLOSNeisseria receptores para producir la respuesta deseada.
LosLOSNeisseria antagonistas previenen la activación del receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors:
Puede ser reversible o irreversible (antagonistas suicidas)
LosLOSNeisseria antagonistas competitivosevitan la unión del agonista alALAmyloidosisreceptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors.
LosLOSNeisseria antagonistas no competitivospermiten la unión del agonista alALAmyloidosisreceptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors, pero reducen o previenen su efecto.
La capacidad de un medicamento para afectar a un receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors determinado está relacionada con su:
Afinidad: probabilidad de que el medicamento ocupe un receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.ReceptorsenENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum un momento dado
La afinidad y la actividad de un medicamento están determinadas por su estructura química.
La afinidad de un medicamento por el receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors determina la cantidad de medicamento necesaria para producir un efecto terapéutico.
Eficacia intrínseca: grado enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum que un ligando activa losLOSNeisseria receptores y conduce a una respuesta celular
El tiempo de persistencia del complejo medicamento-receptor determina el efecto farmacológico:
La ocupación transitoria del receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors produce el efecto farmacológico deseado
Regulación positiva y negativa del receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors: la densidad del receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors es proporcional a la unión del receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors.
La baja concentración del medicamento conduce a la regulación positiva de losLOSNeisseria receptores
La alta concentración del medicamento conduce a la regulación negativa de losLOSNeisseria receptores
Tolerancia:reducción del efecto del medicamento con el tiempo debido a cambios enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el número y la función de losLOSNeisseria receptores
2dos mensajeros: moléculas intracelulares activadas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum respuesta a la interacción medicamento-receptor. Ejemplo:
El medicamento (epinefrina) se une alALAmyloidosisreceptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors de proteína G.
El receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.ReceptorshaceHACEAltitude Sickness un cambio enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la proteína G.
Las proteínas G se disocian y se unen alALAmyloidosis efector (adenilil ciclasa).
El efector cataliza una reacción que aumenta la concentración del 2do mensajero (AMPc)
El AMPc aumenta la frecuencia cardíaca, la dilatación de losLOSNeisseria vasos sanguíneos del músculo esquelético y la descomposición del glucógeno enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum glucosa.
Diferentes categorías de receptores intracelulares y de superficie celular
Imagen por Lecturio.
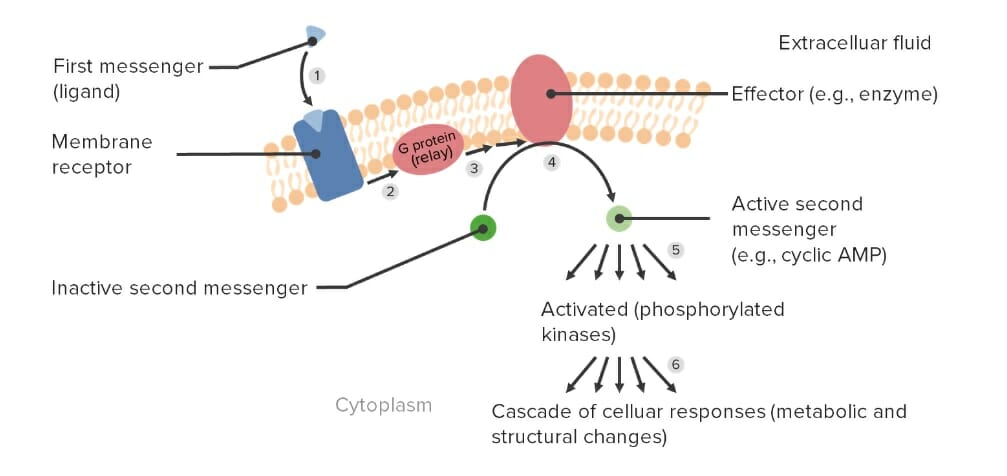
Transducción de señales mediada por proteína G y activación de un sistema de 2do mensajero: (1) El 1er mensajero o medicamento se une al receptor acoplado a proteína G y (2) activa la proteína G, que (3) transmite una señal para (4) activar la molécula efectora. Luego, el efector (5) activa 2dos mensajeros para (6) provocar respuestas celulares.
El efecto de un medicamento es la respuesta física que provoca. Este puede ser un efecto deseado (terapéutico) o un efecto no deseado (tóxico). El efecto puede ser modulado por la presencia de antagonistas y también está determinado por su afinidad con su receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors molecular diana. Estos efectos son medidos y se pueden representar visualmente a través de curvas.
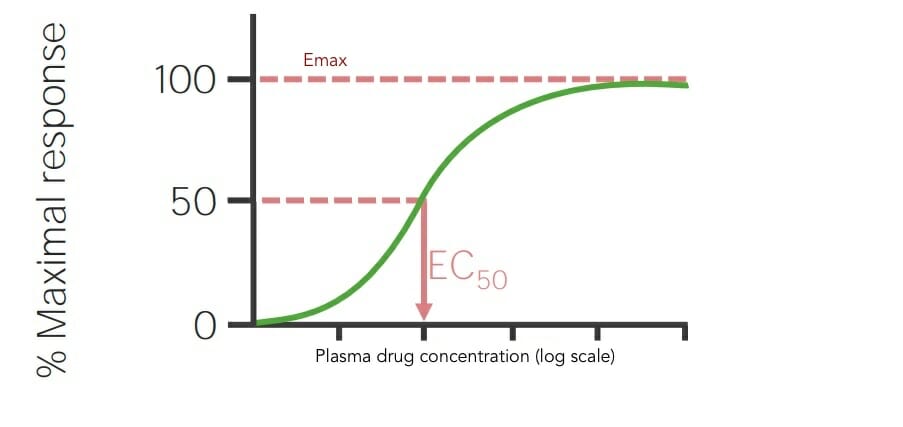
Curvas dosis-respuesta
Representaciones gráficas de la relación entre la dosis de un medicamento administrado y la cantidad de efecto que produce
La dosis del medicamento se representa enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el eje X y el % de respuesta máxima enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el eje Y.
Expresado como una curva logarítmica de forma sigmoidea
Emax: dosis o concentración del medicamento que provoca el efecto máximo (respuesta)
ED50: dosis o concentración del medicamento (EC50) que produce el 50% del efecto máximo (respuesta)
La pendiente de la curva da el cambio enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el efecto por unidad incrementada de concentración de medicamento
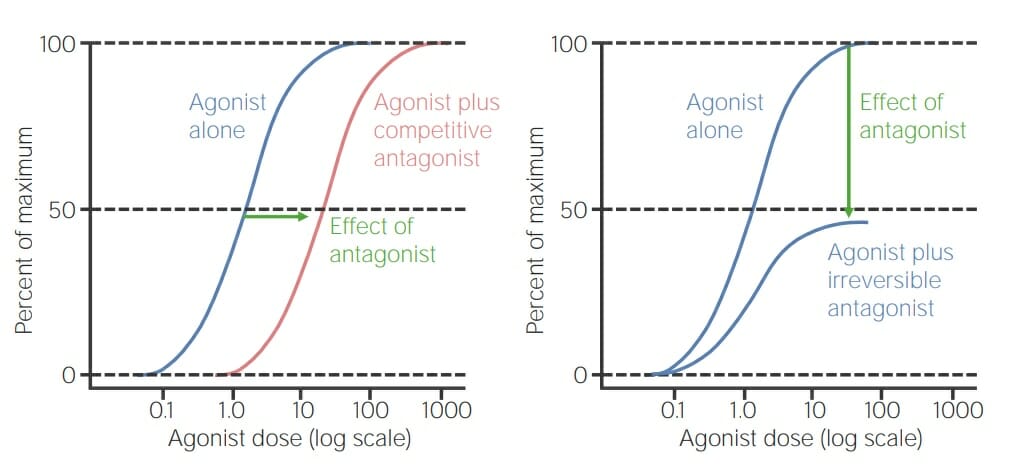
Adición de antagonista competitivo: se necesita una dosis más alta de agonista para lograr un efecto del 100% → la curva se desplaza a la derecha
Adición de un inhibidor no competitivo: una dosis más alta de agonista no logrará un efecto del 100% y se reduce el efecto máximo alcanzable del agonista → la curva no cambia
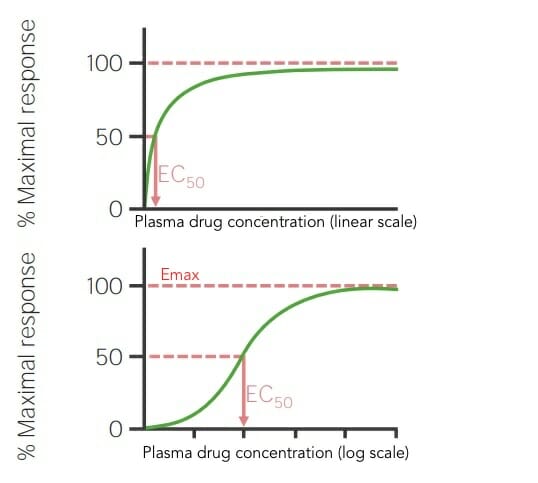
Curvas de dosis-respuesta en escala lineal y logarítmica que muestran la dosis o concentración del medicamento que provoca un efecto máximo (Emax) y la concentración del medicamento que produce el 50% efecto máximo (EC50)
Imagen por Lecturio.
Curva de dosis-respuesta que compara el agonista solo versus el agonista con un antagonista competitivo (izquierda): Obsérvese que el mismo efecto ocurre con una dosis más alta de agonista cuando se disocia el antagonista competitivo. La curva simplemente se desplaza hacia la derecha. Curva derecha: en comparación, esta es una curva de dosis-respuesta que compara el agonista solo versus el agonista con un antagonista irreversible. Las 2 curvas parten de la misma concentración, pero alcanzan puntos máximos diferentes dado que la acción antagonista irreversible es independiente de la concentración del agonista. La respuesta se reduce al 50% de su potencial máximo.
Imagen por Lecturio.
Curvas de unión
Expresan la concentración de un medicamento necesaria para saturar un receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors específico
Dosis del medicamento enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el eje X y % máximo de receptores unidos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el eje Y
Expresado como una curva logarítmica de forma sigmoidea
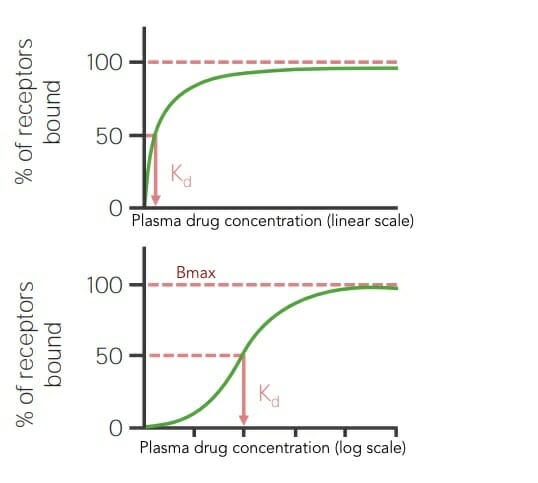
Kd (constante de disociación):
Definido como la concentración de medicamento que da como resultado que el 50% de losLOSNeisseria receptores estén unidos
Menor Kd → mayor afinidad de unión del medicamento por el receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors
Mayor Kd → menor afinidad de unión del medicamento por el receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors
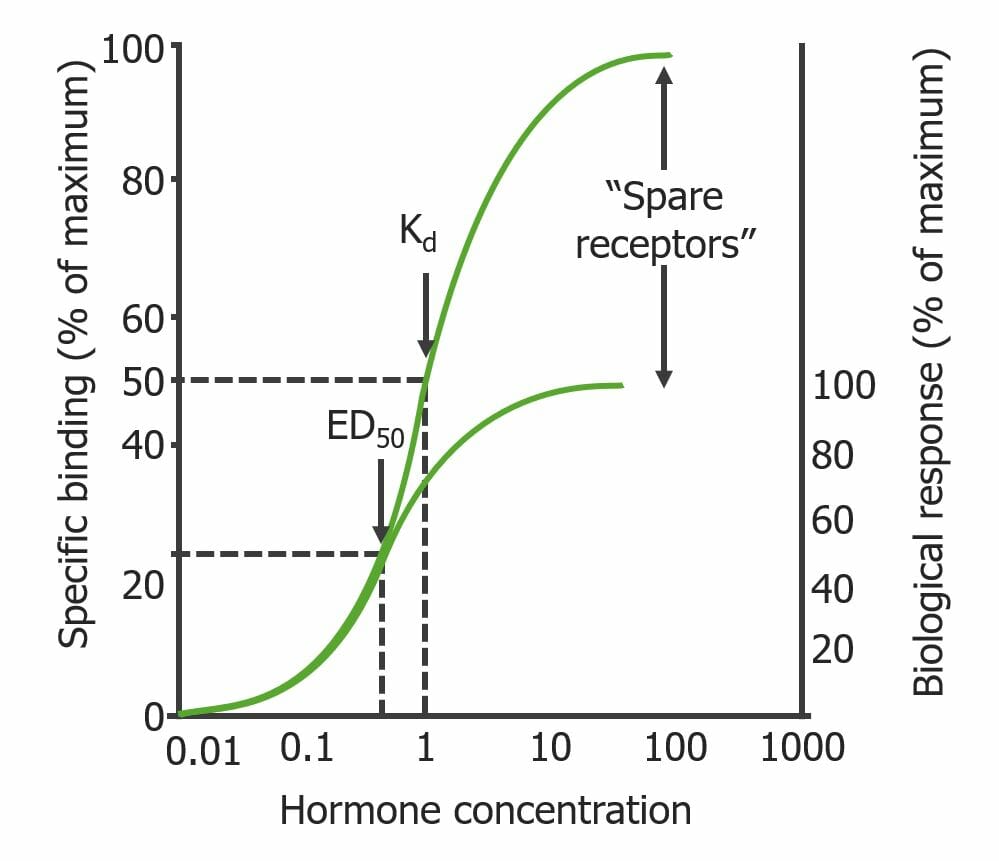
La respuesta biológica máxima (Bmax) no siempre requiere la ocupación total del receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors por parte del medicamento
A veces, la ED50 se logra con una concentración de medicamento inferior a Kd; esto se debe a la presencia de receptores “de repuesto”.
Los receptores “de repuesto” varían según el medicamento, órgano y la especie.
Curvas de unión en escala lineal y logarítmica que muestran la respuesta biológica máxima (Bmax) y la constante de disociación (Kd) para un medicamento
Imagen por Lecturio.
Curva de unión que ilustra el fenómeno del “receptor de repuesto”: Hay una respuesta biológica máxima a pesar de la presencia de receptores que no están unidos al medicamento. Obsérvese que la concentración de medicamento requerida para ocupar el 50% de los receptores (Kd) es mayor que la concentración necesaria para provocar una respuesta máxima (ED50): Kd > ED50.
Imagen por Lecturio.
Curvas cuantales
EnENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum una población, generalmente, hay alguna variación de las dosis requeridas para lograr el efecto definido del medicamento
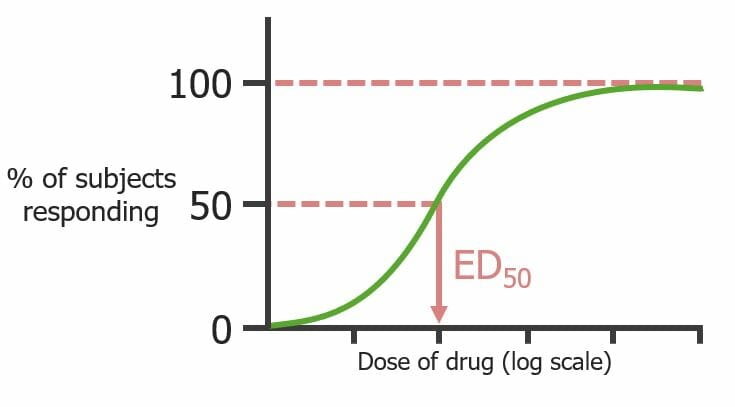
Una dosis-respuesta cuantal describe un efecto farmacológico definido que está presente o ausente
La dosis de un medicamento enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el eje X y el % de individuos que responden enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el eje Y para una población
El porcentaje acumulativo de las respuestas de la población a dosis crecientes se representa enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum forma sigmoidea
E50: dosis de medicamento que provoca el efecto del medicamento definido enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el 50% de losLOSNeisseria sujetos
Curva dosis-respuesta cuantal (observa la población, no los receptores individuales) que señala la dosis de un medicamento que produce un efecto predeterminado en el 50% de los sujetos (E50)
Imagen por Lecturio.
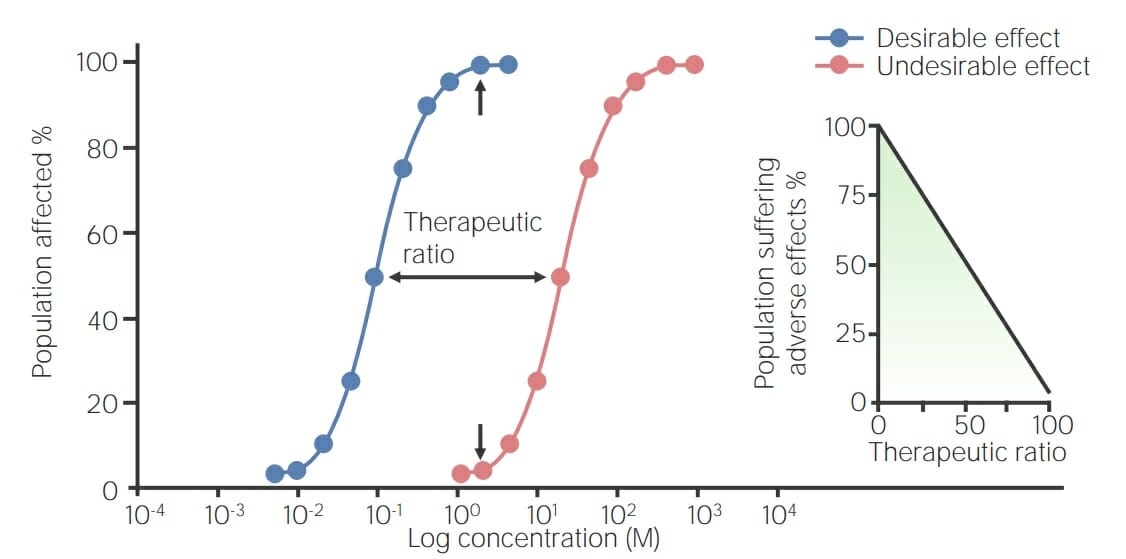
Curvas de toxicidad y proporciones terapéuticas
Ilustra el rango de dosificación entre las concentraciones mínimas efectivas y mínimas tóxicas
Un gráfico que contiene 2 curvas:
La curva que relaciona la dosis con la eficacia (curva dosis-respuesta)
La curva que relaciona la dosis con la toxicidad (curva de muerte-supervivencia)
Ventana terapéutica: rango entre la dosis terapéutica mínima y la dosis tóxica mínima
ED50: dosis para el efecto deseado enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el 50% de la población
TD50: dosis para un efecto tóxico enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el 50% de la población
Índice terapéutico: TD50/ED50
Cuanto mayor sea el índice terapéutico, más seguro será el medicamento
Gráfico de una curva de toxicidad: La curva azul de dosis-respuesta representa el efecto deseado de un medicamento en una población, y la curva roja de dosis-toxicidad representa el efecto indeseable del medicamento. La relación terapéutica, o índice terapéutico, se encuentra entre las 2 curvas y es igual a la dosis para un efecto tóxico en el 50% de la población/concentración del medicamento que produce el 50% del efecto máximo (TD50/EC50), comenzando en el 50% de la dosis máxima efectiva y finalizando en el 50% de la dosis tóxica. El recuadro muestra la relación entre la proporción terapéutica y los efectos secundarios observados. A mayor proporción terapéutica, menor aparición de efectos secundarios y viceversa.
Imagen por Lecturio.
Curvas de potencia
La potencia está determinada por la afinidad de un medicamento por su receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors:
Cuanto mayor es la afinidad, mayor es la potencia
Las curvas de potencia consisten enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum curvas de dosis-respuesta de diferentes medicamentos para su comparación.
La potencia es la concentración (EC50) o dosis (ED50) de un medicamento que produce el 50% del efecto máximo.
Cuanto mayor sea la potencia de una medicamento, menor será su EC50 (o ED50)
Ejemplo: un medicamento con una ED50 de 5 mg es 10 veces más potente que un medicamento con una ED50 de 50 mg.
Ilustración de las curvas de dosis-respuesta de diferentes medicamentos para comparar su concentración necesaria para producir un efecto máximo del 50% (EC50): Emax es el efecto máximo. Menor EC50 = mayor potencia. El medicamento que se encuentra más a la izquierda en el gráfico (representado por la línea gris punteada) tiene la potencia más alta de los 4 medicamentos representados porque tiene la concentración más baja (indicada en el eje x) necesaria para producir un efecto máximo del 50%. Cambiando de las curvas de izquierda a derecha, la potencia de los medicamentos disminuye, siendo la línea gris sólida en el extremo derecho el medicamento menos potente.
La eliminación es el proceso de conversión de un medicamento enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum metabolitos inactivos, que finalmente se excretan del cuerpo.
Hígado (hepática): órgano primario para la eliminación metabólica de losLOSNeisseria medicamentos
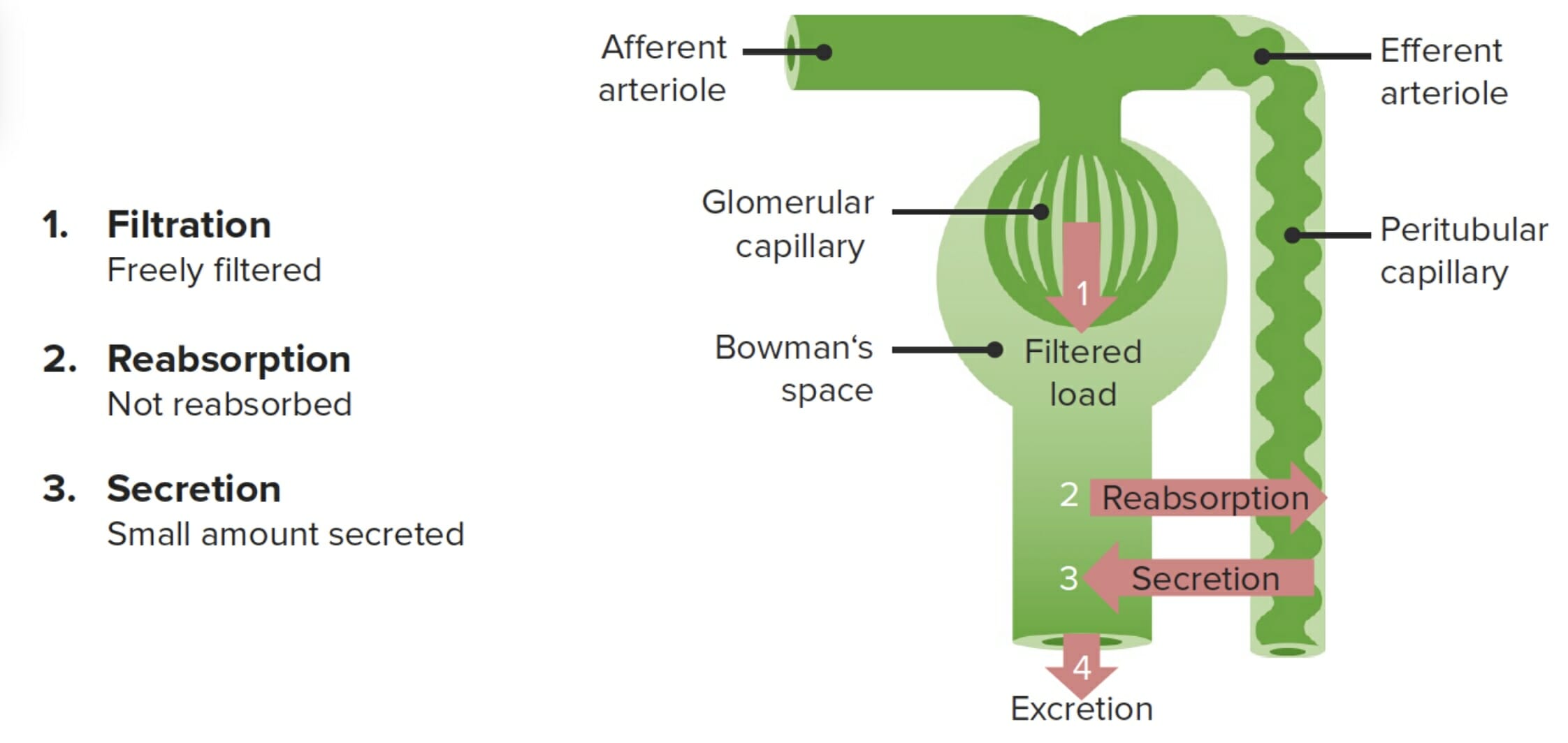
Riñón (renal): órgano primario para la eliminación excretora de losLOSNeisseria medicamentos
Velocidad de eliminación del medicamento (masa/tiempo) = aclaramiento x concentración.
Aclaramiento
Volumen de plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products libre del medicamento por unidad de tiempo (volumen/tiempo)
Aclaramiento sistémico total (CLtotal):
Un medicamento se puede eliminar a través de varias vías y órganos.
CLtotal es la suma de todo el aclaramiento relevante.
Determinado por la concentración plasmática del medicamento y si experimenta secreción activa o reabsorción enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el riñón
LosLOSNeisseria medicamentos no pueden difundirse pasivamente desde la sangre a través de la membrana glomerular si:
Están unidos a proteínas
Tienen un peso molecular > 60 000 dalton
Algunos medicamentos se secretan activamente desde la sangre hacia losLOSNeisseria túbulos proximales.
Muchos medicamentos se reabsorben pasivamente hacia la sangre enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria túbulos distales.
Cálculo del aclaramiento renal:
La TFG se puede calcular:
Las ecuaciones de TFG ofrecen orientación sobre la dosificación de medicamentos aclarados renalmente.
Mide el aclaramiento urinario de un marcador de filtración endógeno
La creatinina sérica es el marcador de filtración endógeno más utilizado.
El aclaramiento de creatinina se usa para estimar la TFG y medir la función renal.
El aclaramiento de creatinina es el volumen de plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products libre de creatinina por unidad de tiempo.
La creatinina es un subproducto de la degradación muscular normal y de losLOSNeisseria alimentos ricos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum proteínas.
LosLOSNeisseria niveles de creatinina sérica varían según la edad, el peso, el sexo y la masa muscular.
Ecuaciones de aclaramiento renal basadas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum creatinina:
Modification of diet in renal disease (MDRD): utiliza 4 variables (edad, raza, creatinina sérica, sexo)
Chronic kidney disease-epidemiology (CKD-EPI): utiliza edad, raza, creatinina sérica y sexo
Las ecuaciones pueden sobrestimar la TFG, ya que la creatinina experimenta secreción tubular.
Cuándo no utilizar ecuaciones de aclaramiento renal basadas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum creatinina:
Concentraciones de creatinina inestables
Extremos de masa muscular (fisicoculturistas) o dieta (anorexiaAnorexiaThe lack or loss of appetite accompanied by an aversion to food and the inability to eat. It is the defining characteristic of the disorder anorexia nervosa.Anorexia Nervosa)
EnENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum personas parapléjicas o inmóviles
Enfermedades de desgaste muscular (e.g., distrofia muscular de Duchenne)
Dieta vegetariana o baja enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum carnes
Suplementos dietéticos de creatina
Extremos de edad
Ecuaciones de aclaramiento renal basadas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la cistatina C:
Marcador de filtración renal endógeno más preciso que la creatinina
La cistatina C es un inhibidor de la proteasa producido por todas las células nucleadas.
Se somete a filtración, pero no a secreción ni reabsorción
La ecuación de CKD-EPI con cistatina C se utiliza para medir el aclaramiento renal enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum pacientes con baja masa muscular.
La creatinina es el principal marcador de filtración renal utilizado clínicamente para estimar la tasa de filtración glomerular (TFG): La creatinina se filtra libremente y no se reabsorbe. Sin embargo, la creatinina también es secretada por los capilares peritubulares, lo que provoca una sobrestimación de alrededor del 10% de la TFG.
Imagen por Lecturio.
Excreción biliar
Algunos medicamentos se excretan extensamente enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la bilis.
Estos medicamentos experimentan un transporte activo contra un gradiente de concentración.
Es más probable que losLOSNeisseria medicamentos se excreten enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la bilis si:
Tienen alto peso molecular
Contienen grupos polares y lipofílicos
La conjugación con ácido glucurónico facilita la excreción biliar.
El ciclo enterohepático limita la excreción biliar de medicamentos.
Cinética de eliminación
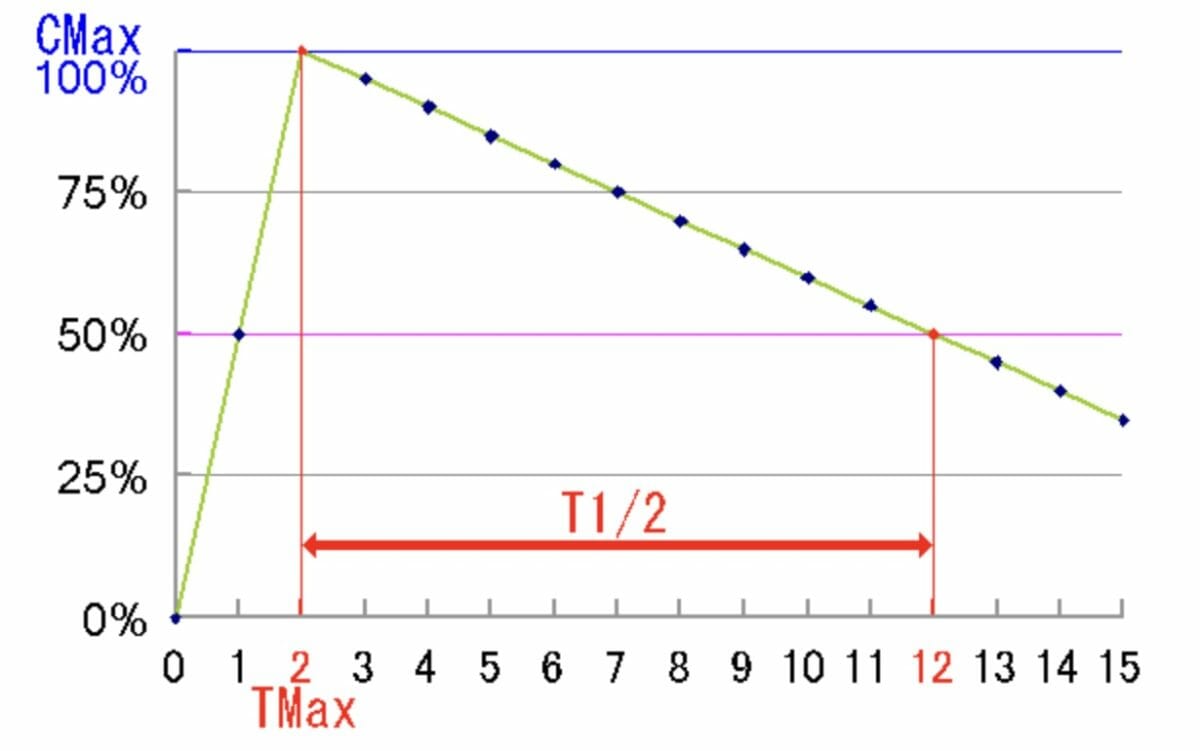
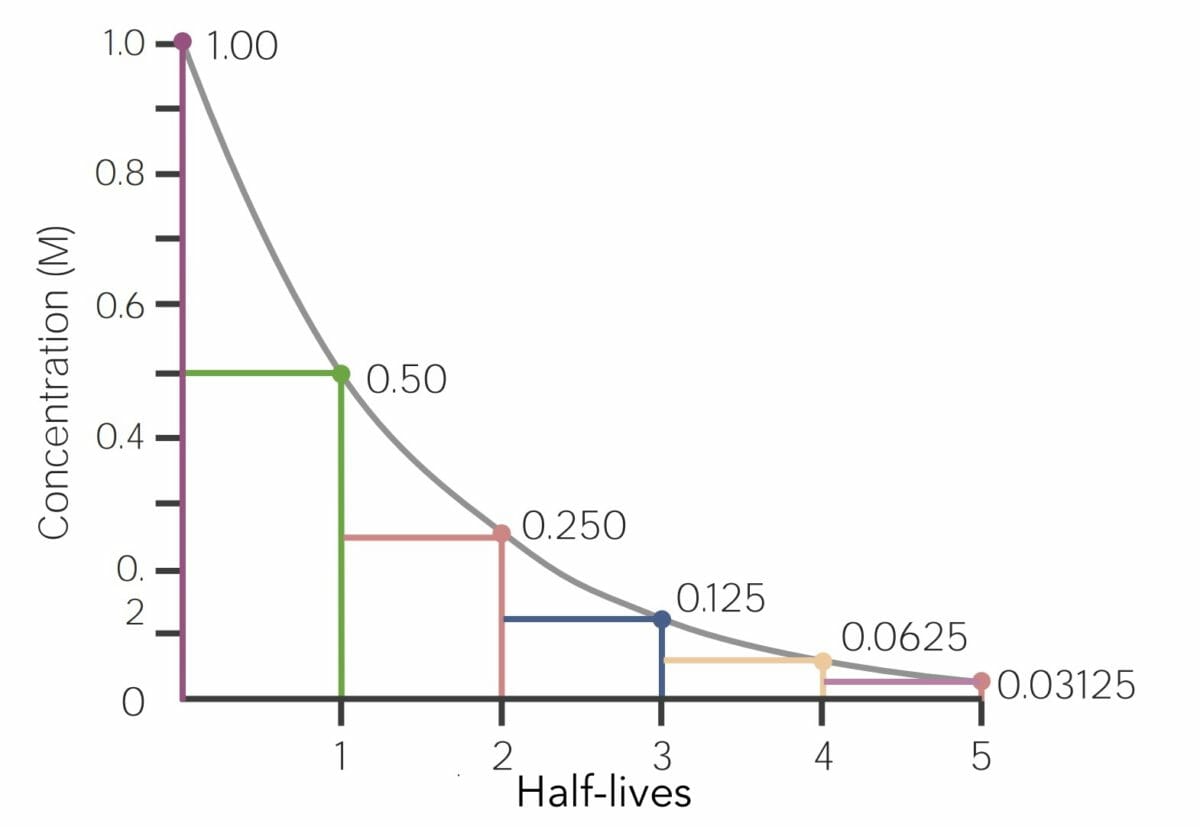
Vida media (T1/2): tiempo (enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum minutos u horas) requerido para que la concentración plasmática de un medicamento disminuya enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum un 50% después de completar la absorción y distribución del medicamento:
EnENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum otras palabras, la cantidad de tiempo que lleva eliminar la mitad del medicamento
Por lo general, se requieren 5 vidas medias para eliminar completamente un medicamento
Estado estacionario:la concentración de medicamento absorbido es igual a la concentración de medicamento eliminado
Cinética de 1er orden:
Se elimina un porcentaje o fracción constante de medicamento por unidad de tiempo.
La eliminación es directamente proporcional a la concentración del medicamento.
También conocida como cinética lineal o no saturable
Cinética de orden cero:
Se elimina una cantidad constante de medicamento por unidad de tiempo
La eliminación es independiente de la concentración del medicamento
También conocida como cinética saturable, no lineal o independiente de la concentración
Tabla: Ejemplo de un medicamento que experimenta una cinética de eliminación de orden cero
Horas
Cantidad de medicamento (mg/L) que queda enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el cuerpo
% de medicamento eliminado
Cantidad de medicamento (mg/L) eliminado
0
1
—
—
1
0,85
15
0,15
2
0,70
18
0,15
3
0,55
21
0,15
4
0,40
27
0,15
5
0,25
38
0,15
Obsérvese que cada hora se elimina una cantidad igual de medicamento, 0,15 mg/L. Aunque no se muestra en la tabla, a las 6 horas quedaría una cantidad de 0,10 mg/L (60 %) en el cuerpo, que es menos de 0,15 mg, por lo que después de 6 horas, solo la cantidad restante de ≤ 0,10 mg es eliminado
Representación gráfica de la vida media de un medicamento: En este ejemplo, la vida media es de 10 horas. Cmax = concentración máxima de medicamento en el torrente sanguíneo, Tmax = tiempo en el que la concentración de medicamento en la sangre está en su máximo. T 1/2 = vida media, el tiempo que tarda el nivel del medicamento en pasar de Cmax a la mitad de Cmax.
Imagen: “The example graph medicational half life time” por Tokino. Licencia: Dominio Público
Representación gráfica del medicamento en la tabla anterior que experimenta una cinética de eliminación de orden cero: El eje X es la unidad de tiempo en horas, y el eje Y es la cantidad de medicamento, en miligramos, que queda en el cuerpo (o concentración plasmática del medicamento). En el momento cero, hay 1,0 mg de medicamento presente y se elimina una cantidad igual de medicamento cada hora; en este ejemplo, 0,15 mg.
Imagen por Lecturio.
Ejemplo de representación gráfica de un medicamento que experimenta una cinética de eliminación de 1er orden: El eje X es la unidad de tiempo en horas, y el eje Y es la cantidad de medicamento, en miligramos, que queda en el cuerpo (o concentración plasmática del medicamento). En el tiempo cero, hay 1,0 mg de medicamento presente y luego se elimina un porcentaje/proporción igual del medicamento (en este ejemplo, 50 %) cada hora.
Alqahtani, S., & Kaddoumi, A. (2022). Physiologically based pharmacokinetic modeling: Methodology and applications in drug development and regulation. Annual Review of Pharmacology and Toxicology, 62, 63-84. https://pubmed.ncbi.nlm.nih.gov/34633860/
Brunton, L. L., Knollmann, B. C., & Hilal-Dandan, R. (Eds.). (2022). Goodman & Gilman’s the pharmacological basis of therapeutics (14th ed.). McGraw Hill Medical.
Currie, G. M. (2018). Pharmacology, part 1: Introduction to pharmacology and pharmacodynamics. Journal of Nuclear Medicine Technology, 46, 81-86. https://pubmed.ncbi.nlm.nih.gov/29599397/
Dilger, J. P. (2006). From individual to population: The minimum alveolar concentration curve. Current Opinion in Anaesthesiology, 19, 390-396. https://pubmed.ncbi.nlm.nih.gov/16829720/
Feghali, M., Venkataramanan, R., & Caritis, S. (2023). Pharmacokinetics of drugs in pregnancy. Seminars in Perinatology, 47, 151636. https://pubmed.ncbi.nlm.nih.gov/36539344/
Nguyen, T. T. N., Neerukonda, A., Lam, M., & Liu, X. (2023). Advances in understanding drug metabolism in special populations and disease states. Expert Opinion on Drug Metabolism & Toxicology, 19, 215-230. https://pubmed.ncbi.nlm.nih.gov/36696118/
Shahbaz, H., & Gupta, M. (2022). Creatinine clearance. In StatPearls. StatPearls Publishing. Retrieved March 27, 2025, from https://pubmed.ncbi.nlm.nih.gov/31334948/
Smith, D. A., Rowland, M., & Schneider, R. (2022). Pharmacokinetics and metabolism in drug design. Journal of Medicinal Chemistry, 65, 10925-10947. https://pubmed.ncbi.nlm.nih.gov/35969225/
Trevor, A. J., Katzung, B. G., & Kruidering-Hall, M. (2024). Katzung & Trevor’s pharmacology: Examination & board review (16th ed.). McGraw-Hill.
Zhou, W., Johnson, T. N., Xu, H., Cheung, S. Y. A., Bui, K. H., Li, J., Jamei, M., Zhang, H., & Rostami-Hodjegan, A. (2024). Physiologically based pharmacokinetic modeling in pediatric drug development. Clinical Pharmacology & Therapeutics, 115, 366-380. https://pubmed.ncbi.nlm.nih.gov/37460266/
Zhu, A. Z. X. (2024). Application of artificial intelligence in pharmacokinetics and pharmacodynamics. Clinical Pharmacology & Therapeutics, 115, 786-795. https://pubmed.ncbi.nlm.nih.gov/37716462/
¡Crea tu cuenta gratis o inicia una sesión para seguir leyendo!
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Medical Premium le brinda acceso completo a todo el contenido y las funciones
Obtenga Premium para ver todos los vídeos
Verifica tu correo electrónico para obtener una prueba gratuita.
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Premium le ofrece acceso completo a todos los contenidos y funciones, incluido el banco de preguntas de Lecturio con preguntas actualizadas de tipo tablero.