


A esclerose lateral amiotrófica (ELA), também conhecida como doença de Lou Gehrig, é uma doença neurodegenerativa esporádica ou hereditária dos neurónios motores superiores (UMNs, pela sigla em inglês) e neurónios motores inferiores (LMNs, pela sigla em inglês). A esclerose lateral amiotrófica é a doença neuronal motora progressiva mais MAIS Androgen Insensitivity Syndrome comum na América do Norte, afetando principalmente homens e indivíduos de etnia caucasiana. Esta doença é caracterizada pela coexistência de sinais e sintomas de UMN e LMN. O diagnóstico é clínico. O tratamento é de suporte e sintomático, progredindo para cuidados paliativos no final da vida.
Last updated: Dec 15, 2025
A causa da ELA esporádica é desconhecida. No entanto, existem vários fatores que contribuem:
O mecanismo patogénico exato da ELA é desconhecido. Parecem existir vias moleculares e genéticas que se combinam para causar apoptose dos UMN e LMN.
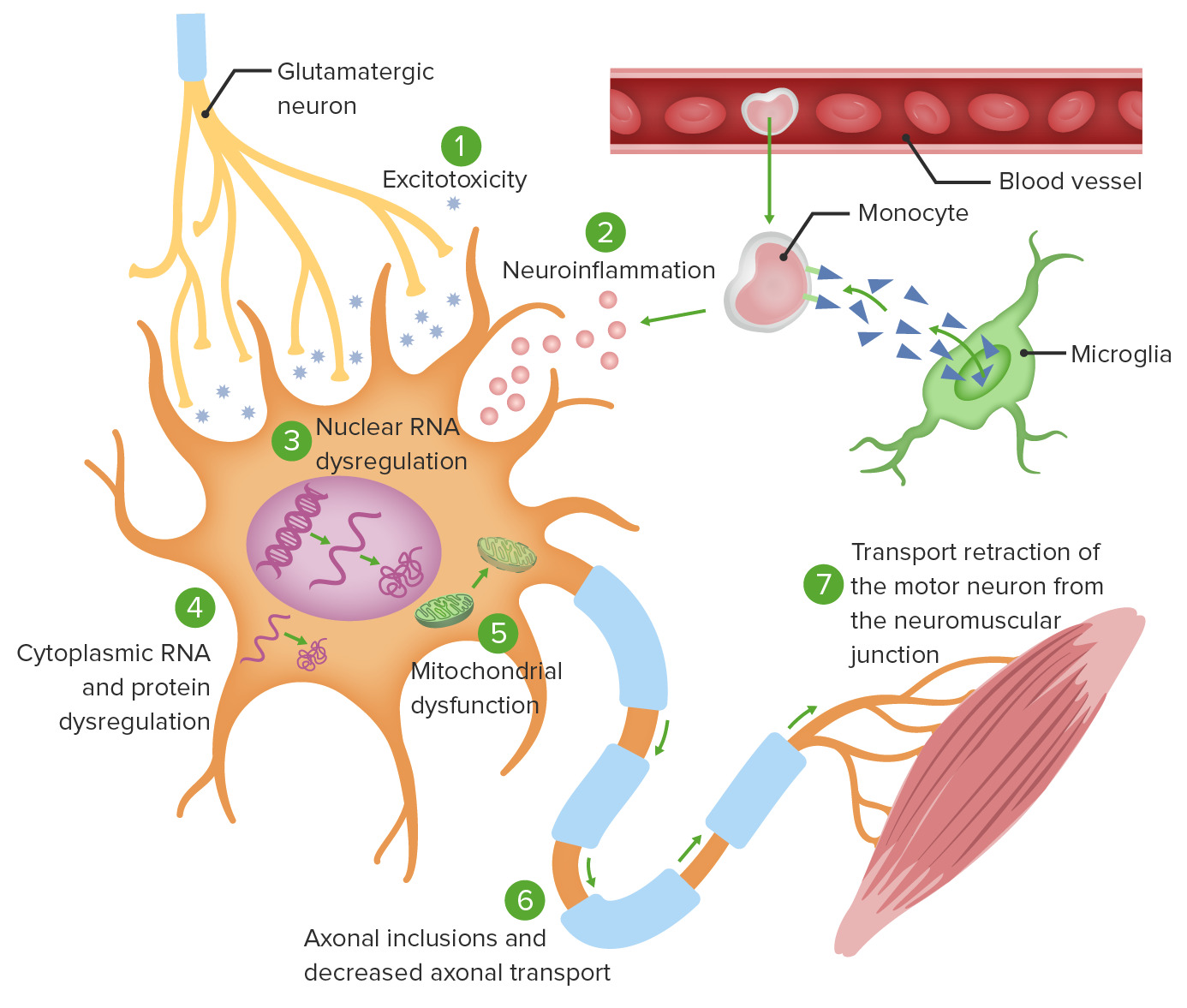
Teorias da fisiopatologia celular da ELA:
1. A excitotoxicidade do glutamato induz processos enzimáticos neurodegenerativos intracelulares.
2. A ativação da microglia mediada por monócitos causa secreção de citocinas pró-inflamatórias.
3. Mutações em genes específicos causam tradução anormal do ARN, levando à agregação de proteínas intranucleares, bem como agregação de proteínas citoplasmáticas (4).
5. Ocorre disfunção mitocondrial, tornando os neurónios incapazes de se adaptar ao stress oxidativo.
6. Os agregados intracelulares prejudicam o transporte axonal.
7. O transporte axonal defeituoso leva à ativação ineficaz da junção neuromuscular alvo.
Durante o curso natural normal da doença, mais MAIS Androgen Insensitivity Syndrome grupos musculares são afetados com o tempo, iniciando-se com uma distribuição assimétrica da fraqueza e tornando-se posteriormente simétrica.
| Sinais do UMN | Sinais do LMN |
|---|---|
| A fraqueza assimétrica (sinal mais MAIS Androgen Insensitivity Syndrome precoce) pode ser atribuída a qualquer um. | |
|
|
O diagnóstico é baseado apenas na apresentação clínica, mas os exames laboratoriais e de imagem são, geralmente, realizados para descartar outras doenças.
Os critérios específicos para o diagnóstico de ELA também são conhecidos como El Escorial World Federation of Neurology criteria El Escorial World Federation of Neurology Criteria Amyotrophic Lateral Sclerosis.
Critérios de inclusão:
Critérios de exclusão:
Usada para descartar outros distúrbios e inclui:
Evidência de desnervação aguda, desnervação crónica e reinervação crónica apoiam o diagnóstico de ELA.
Sem tratamento definitivo. O tratamento atual é sintomático e de suporte.
A terapêutica médica é baseada nas condições subjacentes e na apresentação clínica.
Nutrição:
Fisioterapia, terapia ocupacional e terapia de comunicação:
O objetivo geral destas terapias é melhorar a capacidade de realizar as atividades da vida diária durante o maior tempo possível. Para isso, são usadas diferentes ferramentas.
| Ferramenta | Propósito | |
|---|---|---|
| Fisioterapia e Terapia ocupacional |
|
|
|
|
|
|
Colar cervical |
Impedir a queda da cabeça |
|
|
|
|
| Terapia de comunicação |
Escrever |
Alternativa à fala à medida que a qualidade vocal diminui |
|
Tabuleiros com o alfabeto |
Alternativa à fala à medida que a qualidade vocal diminui e a escrita fica prejudicada |
|
|
Dispositivos eletrónicos de comunicação assistida |
Dispositivos de assistência por voz à medida que a qualidade vocal diminui |
