La esclerosis lateral amiotrófica (ELA), también conocida como enfermedad de Lou Gehrig, es una enfermedad neurodegenerativa hereditaria o esporádica de las neuronas motoras superiores y las neuronas motoras inferiores. La esclerosis lateral amiotrófica es la enfermedad progresiva más frecuente de las neuronas motoras en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum América del Norte afectando principalmente a hombres e individuos de etnia caucásica. Esta enfermedad se caracteriza por la coexistencia de signos y síntomas de las neuronas motoras superiores e inferiores. El diagnóstico se realiza clínicamente. El tratamiento es de soporte y sintomático, progresando hacia los LOS Neisseria cuidados al AL Amyloidosis final de la vida.
Last updated: Dec 15, 2025
Se desconoce la causa de la ELA esporádica. Sin embargo, hay múltiples factores predisponentes:
Se desconoce el mecanismo patogénico exacto de la ELA. Parece haber vías tanto moleculares como genéticas que se combinan para causar la apoptosis Apoptosis A regulated cell death mechanism characterized by distinctive morphologic changes in the nucleus and cytoplasm, including the endonucleolytic cleavage of genomic DNA, at regularly spaced, internucleosomal sites, I.e., DNA fragmentation. It is genetically-programmed and serves as a balance to mitosis in regulating the size of animal tissues and in mediating pathologic processes associated with tumor growth. Ischemic Cell Damage de las neuronas motoras superiores e inferiores.
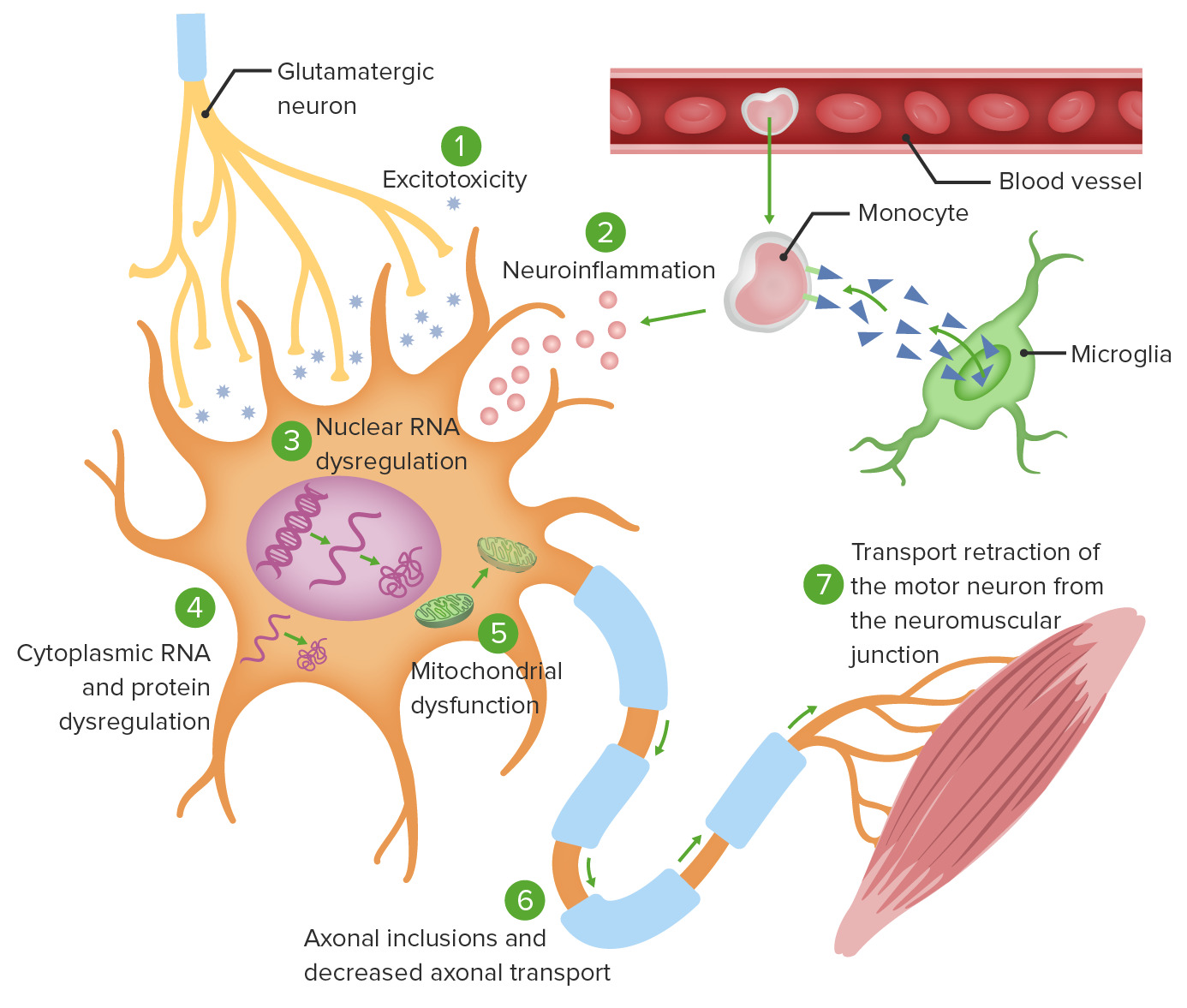
Teorías de la fisiopatología celular de la ELA:
1. La excitotoxicidad del glutamato induce procesos enzimáticos neurodegenerativos intracelulares.
2. La activación de la microglía mediada por monocitos provoca la secreción de citocinas proinflamatorias.
3. Las mutaciones genéticas específicas causan una traducción anormal del ARN, lo que conduce a la agregación de proteínas intranucleares, así como a la agregación de proteínas citoplasmáticas (4).
5. Se produce una disfunción mitocondrial, lo que hace que las neuronas sean incapaces de adaptarse al estrés oxidativo.
6. Los agregados intracelulares alteran el transporte axonal.
7. El transporte axonal defectuoso provoca una activación ineficaz de la unión neuromuscular meta
Durante el curso natural de la enfermedad, más grupos musculares se ven afectados con el tiempo, comenzando con una distribución asimétrica de la debilidad y luego volviéndose simétrica.
| Signos de la neurona motora superior | Signos de la neurona motora inferior |
|---|---|
| La debilidad asimétrica (signo más precoz) se puede atribuir a cualquiera de las dos. | |
|
|
El diagnóstico se realiza solo con la presentación clínica, pero las pruebas de laboratorio e imagenología generalmente se realizan para descartar otras enfermedades.
Los LOS Neisseria criterios específicos para el diagnóstico de la ELA también se conocen como criterios de Gold Coast.
Se utilizan para descartar otros trastornos e incluyen:
Evidencia de denervación aguda, denervación crónica y reinervación crónica que respalda el diagnóstico de la ELA.
No hay tratamiento definitivo. El tratamiento actual es sintomático y de soporte.
La terapia médica se basa en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum las condiciones subyacentes y presentación clínica.
Nutrición:
Terapia física, terapia ocupacional y terapia de lenguaje:
El objetivo general de estas terapias es mejorar la capacidad de realizar actividades de la vida diaria durante el mayor tiempo posible. Para ello se utilizan distintas herramientas.
| Herramienta | Propósito | |
|---|---|---|
| Terapia física y Terapia ocupacional |
|
|
|
|
|
|
Collarete cervical |
Prevenir la caída de la cabeza |
|
|
|
|
| Terapia de lenguaje |
Escritura |
Alternativa al AL Amyloidosis habla cuando disminuye la calidad vocal |
|
Tableros alfabéticos |
Alternativa al AL Amyloidosis habla a medida que disminuye la calidad vocal y se deteriora la escritura. |
|
|
Dispositivos electrónicos de ayuda a la comunicación |
Dispositivo de asistencia vocal ante el deterioro de la calidad vocal |