


A doença de Huntington (DH) é uma doença neurodegenerativa progressiva com um modo de hereditariedade autossómico dominante e mau prognóstico. É causada pelas repetições do trinucleotídeo citosina-adenina-guanina (CAG) no gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics da Huntingtina ( HTT HTT Huntington Disease). A apresentação clínica mais MAIS Androgen Insensitivity Syndrome comum na idade adulta é um distúrbio do movimento conhecido como coreia: movimentos abruptos e involuntários da face, tronco e membros. Os aspectos psiquiátricos e cognitivos também são característicos, e os doentes com DH correm um risco maior de suicídio durante o curso da doença. O diagnóstico é primariamente clínico, frequentemente com história familiar positiva seguida de confirmação genética. O tratamento por equipas interdisciplinares é de suporte, com o objetivo de manter a qualidade de vida. Em doentes com doença de Huntington o tratamento da depressão, agitação e psicose é prioritário em relação ao tratamento da coreia.
Last updated: Dec 27, 2025
A doença de Huntington (DH) é uma doença neurodegenerativa hereditária e progressiva que causa um distúrbio coreiforme do movimento, marcha instável e depressão.
A doença de Huntington é causada por uma alteração genética autossómica dominante no gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics da huntingtina ( HTT HTT Huntington Disease) presente no cromossoma 4p.
A DH é causada por repetições de trinucleotídeos CAG no gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics HTT HTT Huntington Disease.
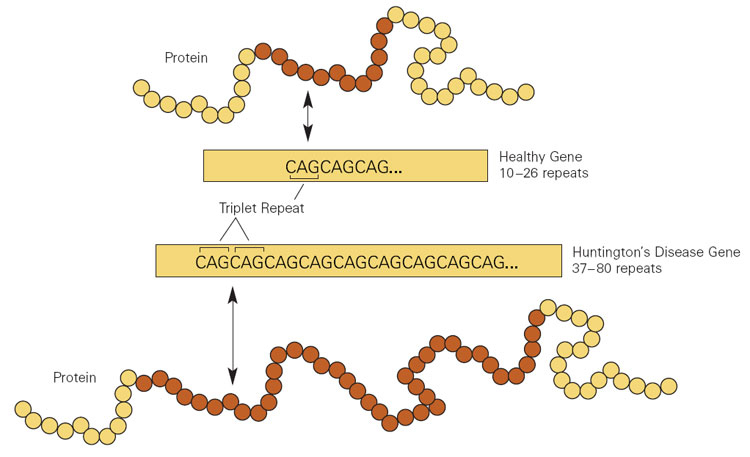
Expansão da repetição da citosina-adenina-guanina (CAG) no gene huntingtina ( HTT), resultando na doença de Huntington
Imagem: “Huntington’s disease patient” por National Institute of Standards and Technology. Licença: Public Domain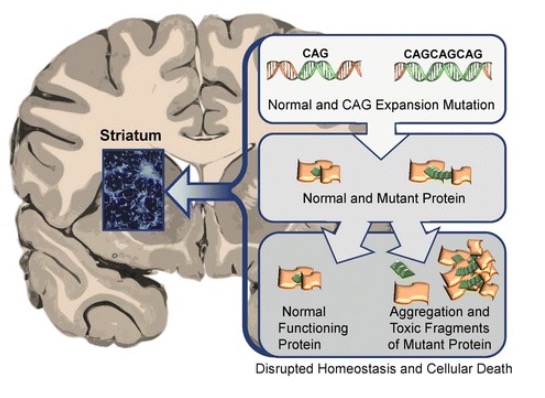
Efeito tóxico das proteínas HTT anormais no estriado na doença de Huntington, levando à neurodegeneração e morte dos neurónios afetados
Imagem: “Neuronal changes in Huntington disease” por Jaenisch R, Zhang F, Gage F. Licença: CC BY 4.0A doença de Huntington afeta várias gerações nas famílias e normalmente estende-se por várias décadas. Os sintomas podem ser motores, cognitivos e comportamentais.
O tratamento da DH inclui suporte médico e psicológico para controlo dos sintomas e manutenção da qualidade de vida. Em pacientes com DH o tratamento da depressão, agitação e psicose é prioritário em relação ao tratamento da coreia.
