La enfermedad de Huntington es un trastorno neurodegenerativo progresivo con un modo de herencia autosómico dominante y de mal pronóstico. Es causada por repeticiones del trinucleótido citosina-adenina-guanina (CAG) en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el gen de la huntingtina ( HTT HTT Huntington Disease). La presentación clínica más común en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la adultez son los LOS Neisseria trastornos del movimiento conocidos como corea: movimientos bruscos e involuntarios de la cara, tronco y extremidades. Las características psiquiátricas y cognitivas también son propias de la enfermedad y los LOS Neisseria pacientes con enfermedad de Huntington tienen un mayor riesgo de suicidio durante el curso de la enfermedad. El diagnóstico es principalmente clínico, a menudo con antecedentes familiares positivos seguidos de confirmación genética. El tratamiento por parte de un equipo interdisciplinario es de soporte, con el objetivo de mantener la calidad de vida. El tratamiento de la depresión, agitación y psicosis es la 1ra prioridad para los LOS Neisseria pacientes con enfermedad de Huntington sobre el tratamiento de la corea.
Last updated: Dec 27, 2025
La enfermedad de Huntington es un trastorno neurodegenerativo hereditario y progresivo que causa trastornos del movimiento coreiformes, marcha inestable y depresión.
La enfermedad de Huntington está causada por una alteración genética autosómica dominante en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el gen de la huntingtina ( HTT HTT Huntington Disease) presente en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el cromosoma 4p.
La enfermedad de Huntington es causada por repeticiones del trinucleótido CAG en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el gen HTT HTT Huntington Disease.
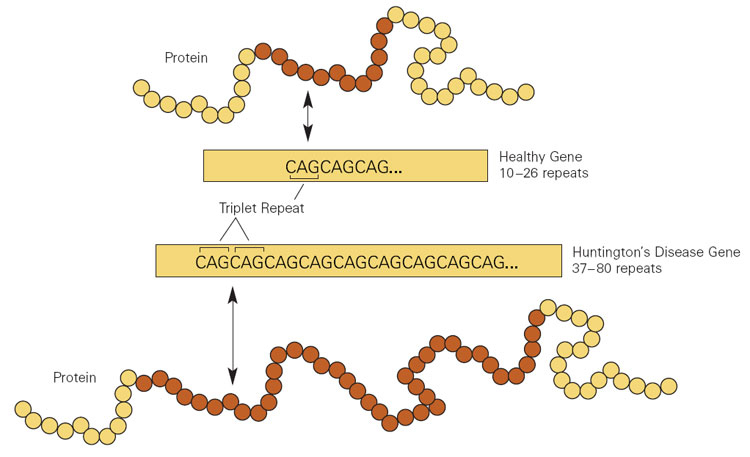
Expansión repetida de citosina-adenina-guanina (CAG) en el gen de la huntingtina (HTT) que da como resultado la enfermedad de Huntington
Imagen: “Huntington’s disease patient” por National Institute of Standards and Technology. Licencia: Dominio Público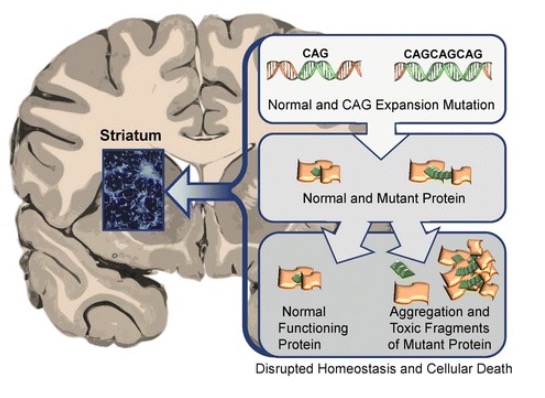
El efecto tóxico de las proteínas HTT anormales en el cuerpo estriado en la enfermedad de Huntington conduce a la neurodegeneración y muerte de las neuronas afectadas
Imagen: “Neuronal changes in Huntington disease” por Jaenisch R, Zhang F, Gage F. Licencia: CC BY 4.0La enfermedad de Huntington afecta a múltiples generaciones de los LOS Neisseria miembros de la familia y, por lo general, se extiende por varias décadas. Los LOS Neisseria síntomas pueden ser motores, cognitivos y conductuales.
El tratamiento de la enfermedad de Huntington incluye soporte médico y psicológico para controlar los LOS Neisseria síntomas y mantener la calidad de vida. El tratamiento de la depresión, agitación y psicosis es la 1ra prioridad para los LOS Neisseria pacientes con enfermedad de Huntington sobre el tratamiento de la corea.