


A deficiência de alfa-1 antitripsina é uma doença genética causada por mutações genéticas hereditárias do gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics SERPINA1, que causam a produção defeituosa do inibidor de protease Protease Enzyme of the human immunodeficiency virus that is required for post-translational cleavage of gag and gag-pol precursor polyproteins into functional products needed for viral assembly. HIV protease is an aspartic protease encoded by the amino terminus of the pol gene. HIV Infection and AIDS alfa-1 antitripsina (AAT). Estas mutações podem levar à deficiência da enzima que causa doença pulmonar, produção de uma forma anormal da enzima que leva à disfunção hepática ou ambos. Os doentes geralmente apresentam enfisema, pneumotórax espontâneo, cirrose, hepatite ou carcinoma hepatocelular. Não há cura conhecida. O tratamento é de suporte e inclui infusão de AAT, tratamento de comorbilidades e transplante de fígado. O prognóstico pode variar de acordo com a forma da doença contraída e a gravidade dos sintomas.
Last updated: Apr 25, 2025
Doença hepática:
Doença pulmonar:
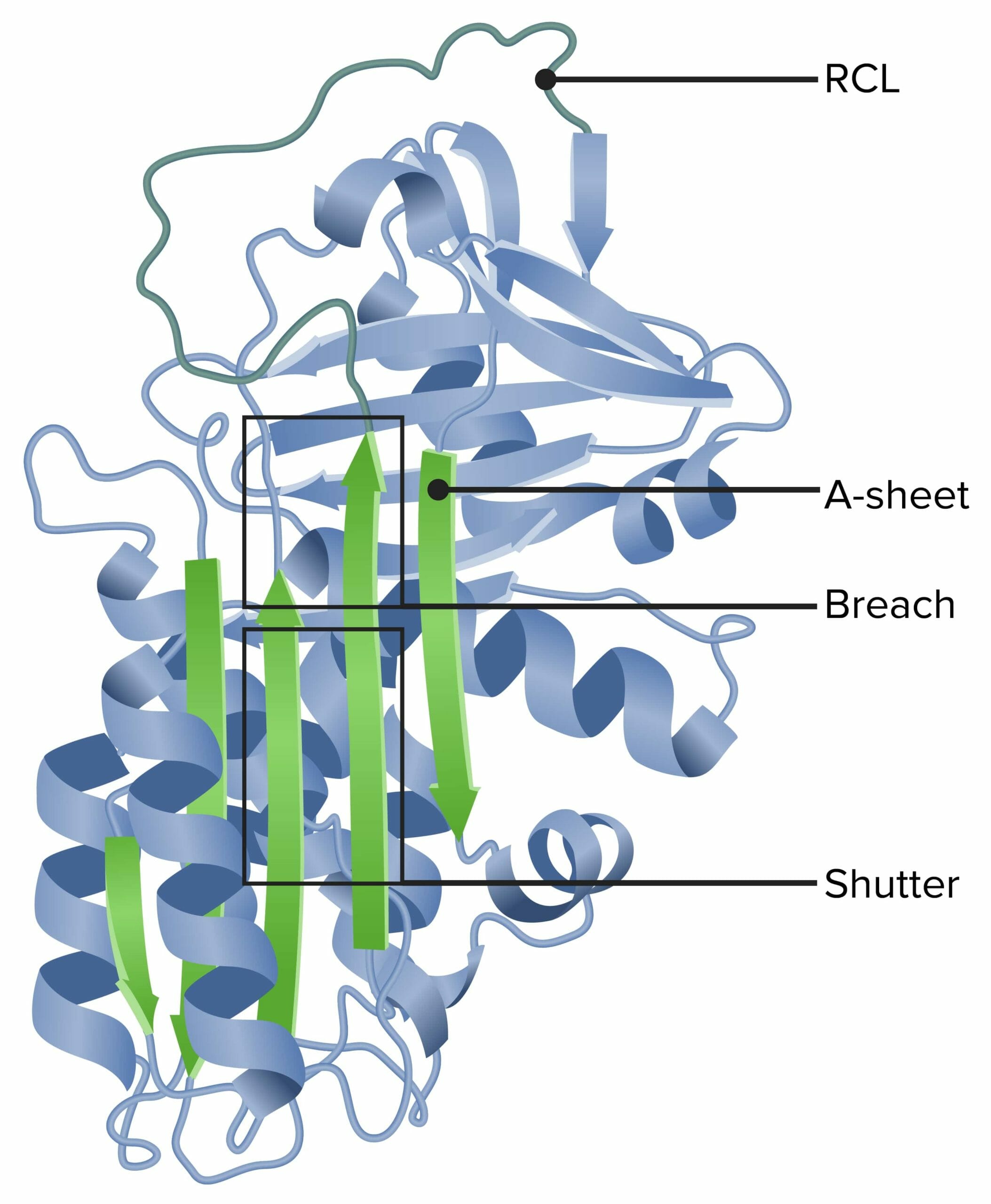
Alfa-1 antitripsina
RCL: loop central reativo
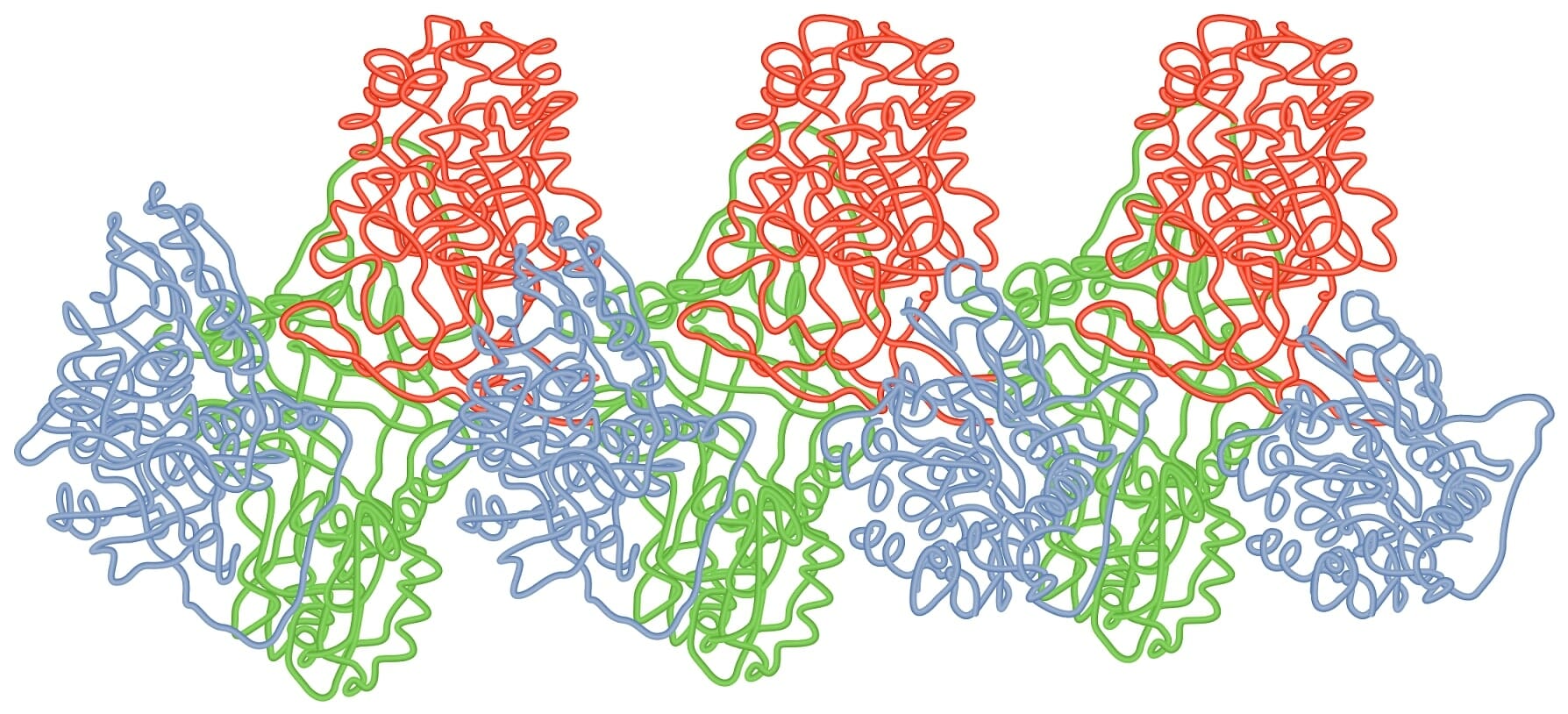
Polímero de folha de loop da moléculas AAT
Imagem por Lecturio.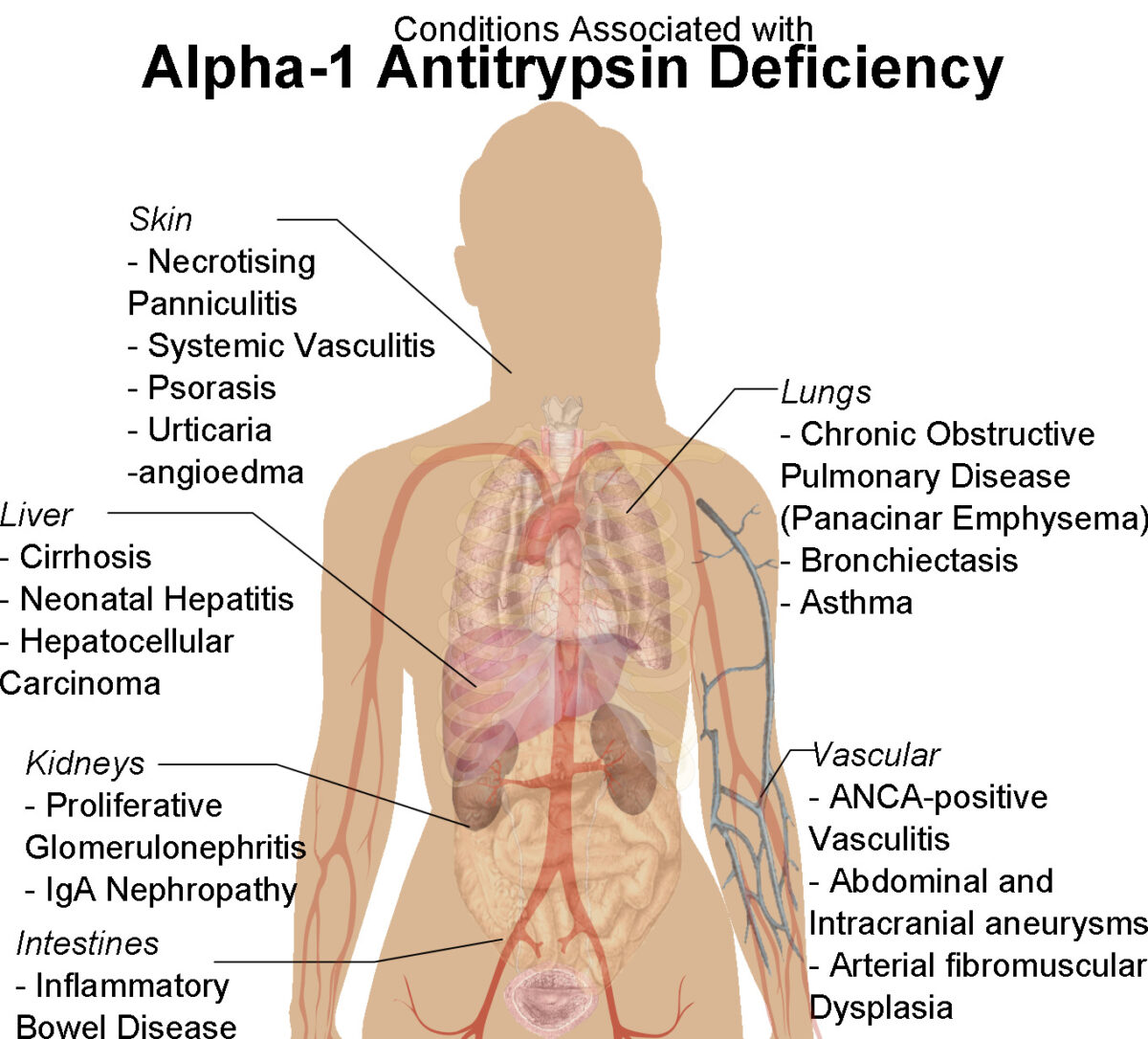
Diagrama de figura feminina humana com condições associadas à deficiência de alfa-1 antitripsina
Imagem : “Human female shadow diagram with conditions associated with Alpha-1 Antitrypsin Deficiency” por Mikael Häggström. Licença: CC0 1.0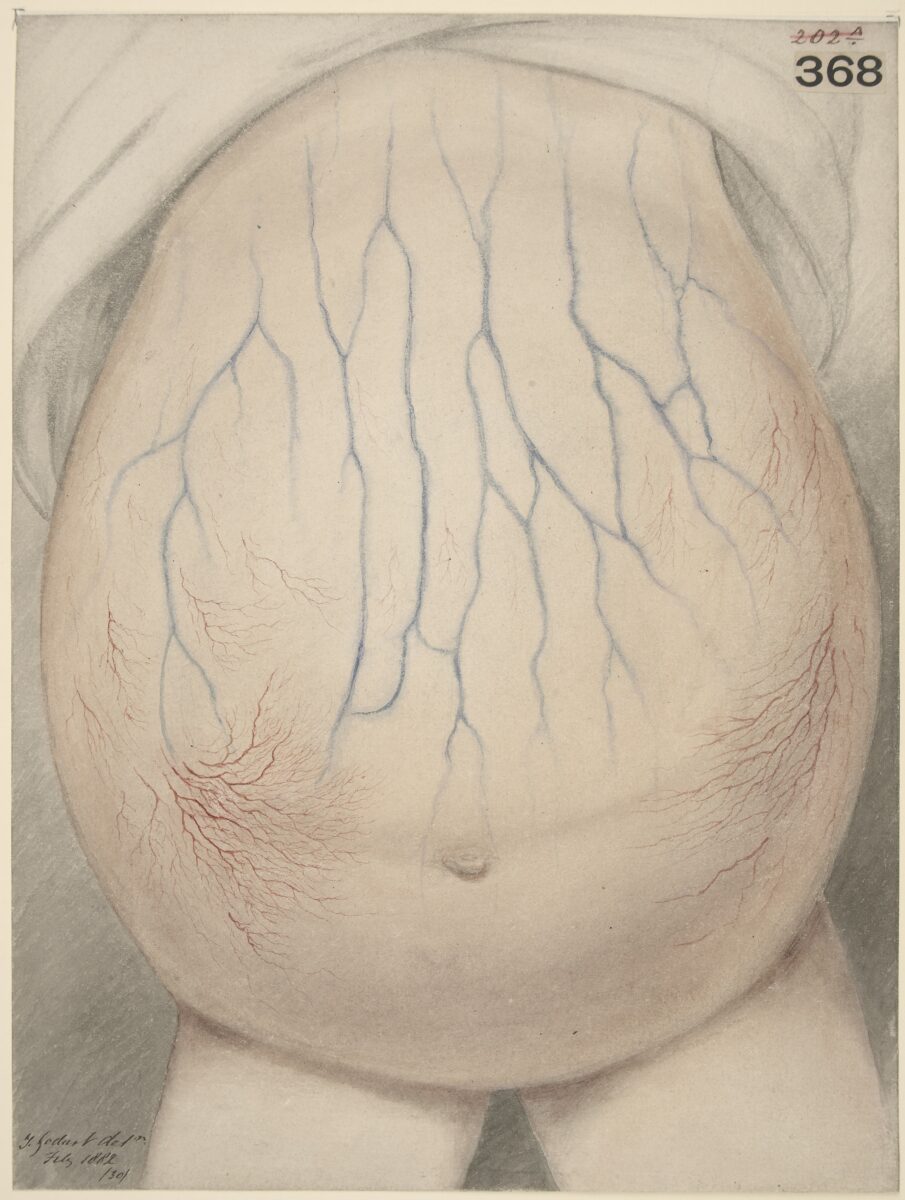
Abdómen de criança afetada com ascite
Imagem : “Watercolour drawing of the abdomen of a child affected with ascites” por galeria Wellcome Collection. Licença: CC BY 4.0