


A deficiência de adesão leucocitária tipo 1 ( LAD1 LAD1 Leukocyte adhesion deficiency type 1 (LAD1) is an inherited condition in which genetic mutations result in a lack of CD18 expression on neutrophils. These mutations lead to a decrease in the ability of neutrophils to migrate from the blood vessels to the site of injury or infection on recruitment, presenting as recurrent infections and delayed wound healing. Leukocyte Adhesion Deficiency Type 1, pela sigla em inglês) é uma doença hereditária na qual existem mutações genéticas que resultam na ausência de expressão de CD18 CD18 Cell-surface glycoprotein beta-chains that are non-covalently linked to specific alpha-chains of the cd11 family of leukocyte-adhesion molecules (receptors, leukocyte-adhesion). A defect in the gene encoding cd18 causes leukocyte-adhesion deficiency syndrome. Leukocyte Adhesion Deficiency Type 1 em neutrófilos. Estas mutações levam a uma diminuição da capacidade dos neutrófilos em migrarem dos vasos sanguíneos para o local da lesão ou infeção ao serem recrutados. Os pacientes com esta patologia terão infeções recorrentes e uma cicatrização mais MAIS Androgen Insensitivity Syndrome lenta das feridas. Na avaliação laboratorial, denota-se uma elevada contagem de neutrófilos. O diagnóstico é confirmado com citometria de fluxo, que demonstra uma deficiência em CD18 CD18 Cell-surface glycoprotein beta-chains that are non-covalently linked to specific alpha-chains of the cd11 family of leukocyte-adhesion molecules (receptors, leukocyte-adhesion). A defect in the gene encoding cd18 causes leukocyte-adhesion deficiency syndrome. Leukocyte Adhesion Deficiency Type 1. O transplante de células estaminais hematopoiéticas é o tratamento de escolha, podendo ser curativo.
Last updated: Dec 15, 2025
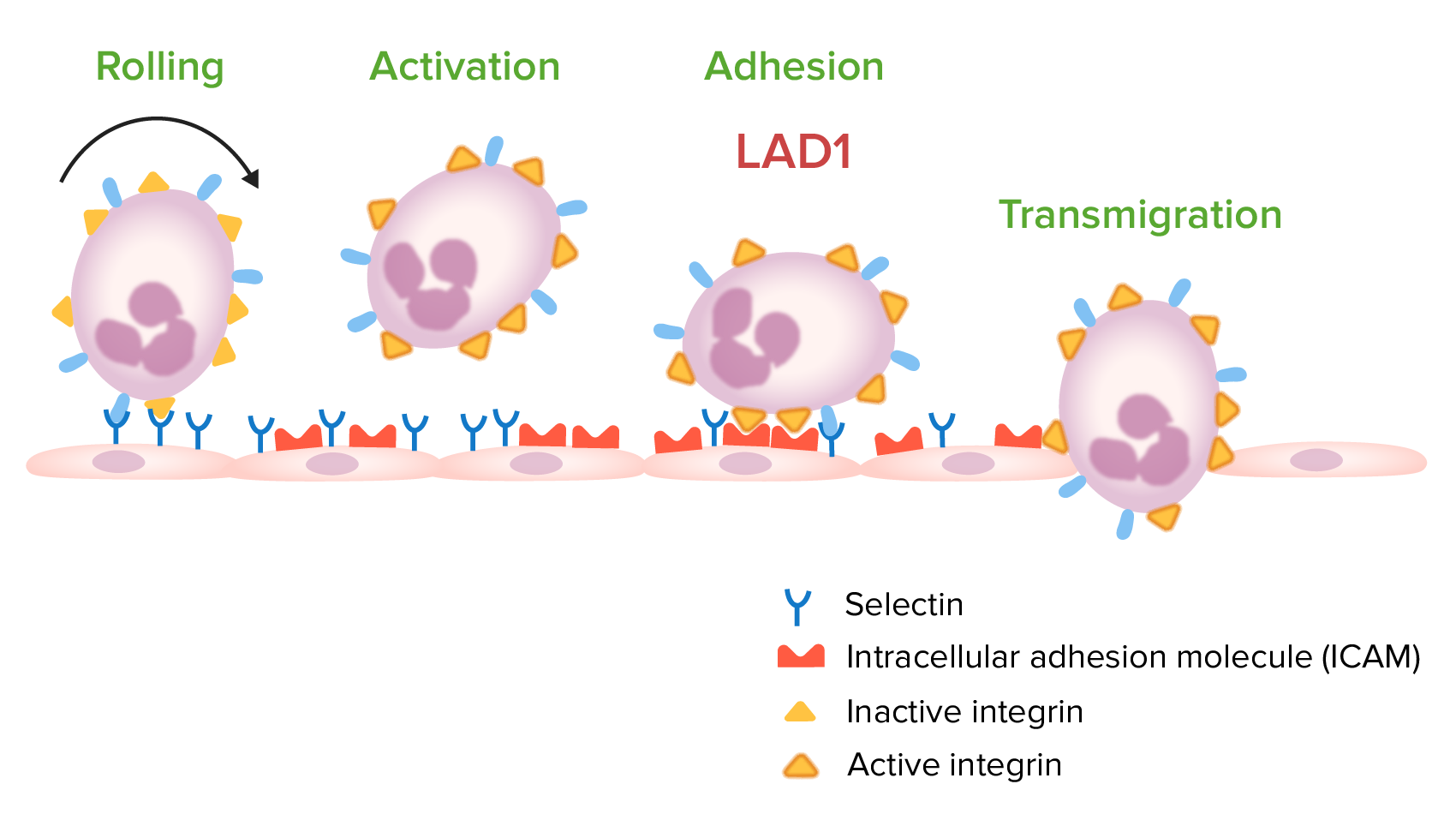
Passos envolvidos na migração de leucócitos:
A deficiência de adesão leucocitária tipo 1 (LAD1, pela sigla em inglês) ocorre devido a uma mutação no gene codificante para CD18. Essa mutação leva a uma deficiência de integrinas ativas utilizadas na adesão de leucócitos ao endotélio vascular, o que impede a transmigração para o local da infeção.
