Os aminoácidos (AAs) podem ser adquiridos através da degradação de proteínas intracelulares ou ingeridas na dieta. Os aminoácidos podem entrar em 3 vias metabólicas dentro do corpo. Estes podem 1) ser reciclados para sintetizar novas proteínas; 2) combinar com cofatores e substâncias para criar derivados de aminoácidos; ou 3) ser catabolizados nos seus grupos funcionais e esqueletos de carbono. Este processo liberta amónio, que entra no ciclo da ureia e produz intermediários para as vias metabólicas energéticas.
Os aminoácidos (AAs) seguem 3 vias metabólicas principais para o seu metabolismo:
Síntese de novas proteínas
Formação de derivados de aminoácidos
Catabolismo de AAs:
O catabolismo consiste na degradação de moléculas complexas em unidades menores para produzir energia ou para serem usadas em reações anabólicas.
Remoção ou troca de grupos funcionais:
Envolve transaminação, desaminação e descarboxilação
Liberta o excesso de nitrogénio na forma de amónio (NH4+), que entra no ciclo da ureia, é convertido em ureia e excretado pela urina
Catabolismo do esqueleto de carbono remanescente:
Em geral, todos os 20 AAs podem ser divididos em 1 de 6 intermediários: piruvato, acetil-CoA, oxaloacetato, alfa-cetoglutarato, succinil-CoA ou fumarato.
Os AAs cetogénicos metabolizam em acetil-CoA, posteriormente usado no ciclo do ácido cítrico, cetogénese ou síntese de ácidos gordos.
Os AAs glicogénicos são convertidos em glicose através da gliconeogénese.
Alguns AAs são glicogénicos e cetogénicos.
Diagrama esquemático do metabolismo dos aminoácidos, incluindo as 3 principais vias: reutilização na síntese de novas proteínas, união com cofatores para produção de derivados de aminoácidos e catabolismo. O catabolismo de aminoácidos inclui a remoção de grupos funcionais e a degradação dos esqueletos de carbono.
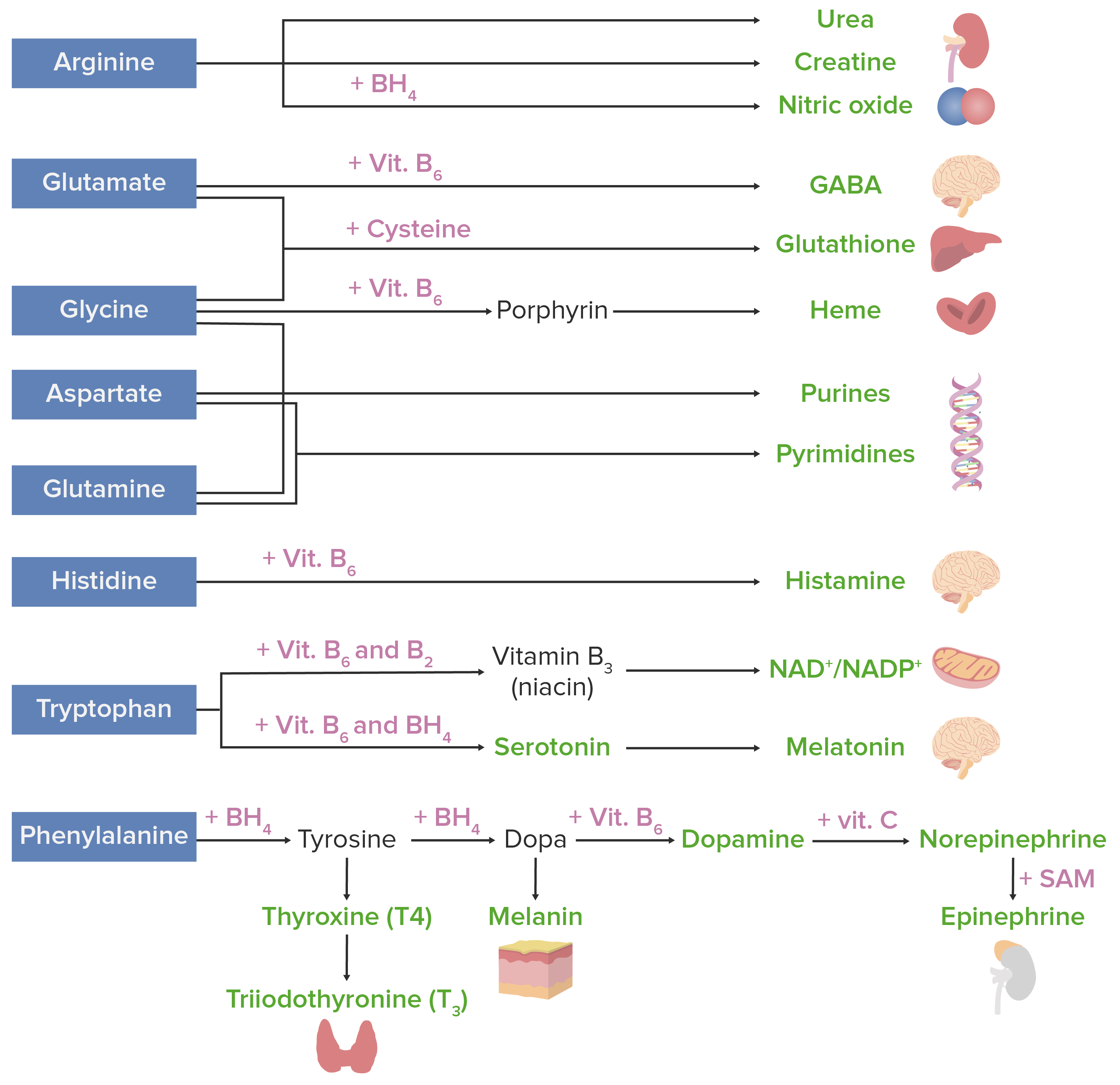
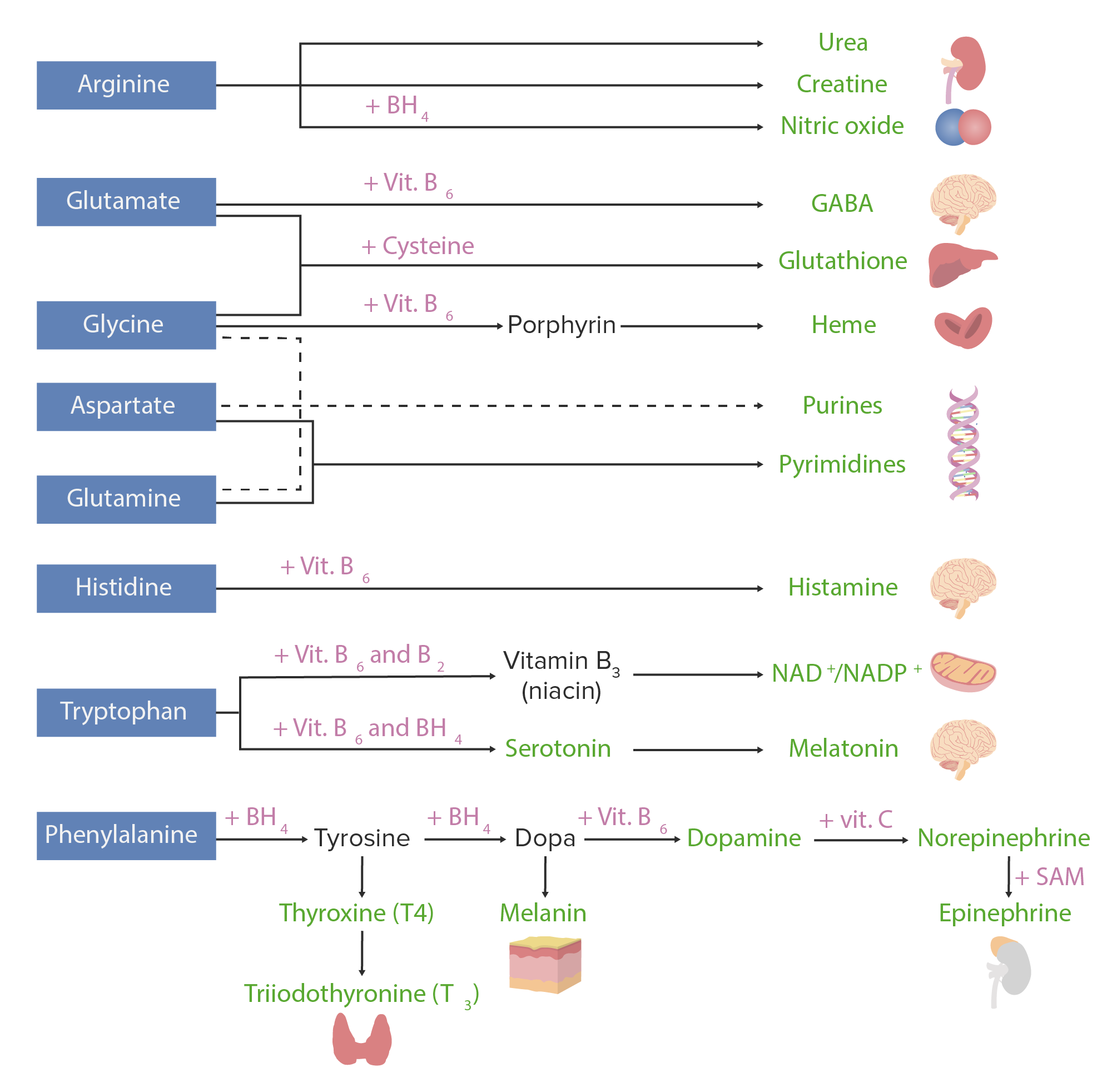
Os aminoácidos podem ser usados para agrupar muitas substâncias. A imagem abaixo mostra as substâncias derivadas de AAAAAmyloidosismaisMAISAndrogen Insensitivity Syndrome importantes em humanos.
Derivados de aminoácidos. Os aminoácidos (a azul) são combinados com certos cofatores ou outros substratos (a rosa) para produzir várias substâncias biologicamente importantes (a verde).
A transaminação é a transferência de um grupo amina de um alfa-AA para um alfa-cetoácido, um AAAAAmyloidosis com um grupo alfa-ceto (=O) em vez de um grupo alfa-amina (NH 2 ).
O AAAAAmyloidosis original perde um grupo amina e ganha um grupo ceto, tornando-se um alfa-cetoácido.
O alfa-cetoácido original perde o seu grupo ceto e ganha um amina, tornando-se um AAAAAmyloidosis não essencial.
A reação é catalisada por enzimas aminotransferases:
Podem ser específicas para um determinado parPARThe PAR is the attributable risk for an entire population. It represents the fraction of cases that would not occur in a population if the exposure was eliminated.Measures of Risk de AAAAAmyloidosis ou um grupo com composições químicas semelhantes
Requerem coenzima piridoxal fosfato (PLP, a forma ativa da vitamina B6)
Encontradas em altas concentrações no fígado
Este processo depende da necessidade. Se houver excesso de um tipo de AAAAAmyloidosis, o grupo amina deste tipo pode ser transferido para fazer outros tipos de AAAAAmyloidosis que o corpo necessita.
Todos os AAs comuns participam da transaminação, exceto lisina, treonina, prolina e hidroxiprolina, que catabolizam via desidrogenase.
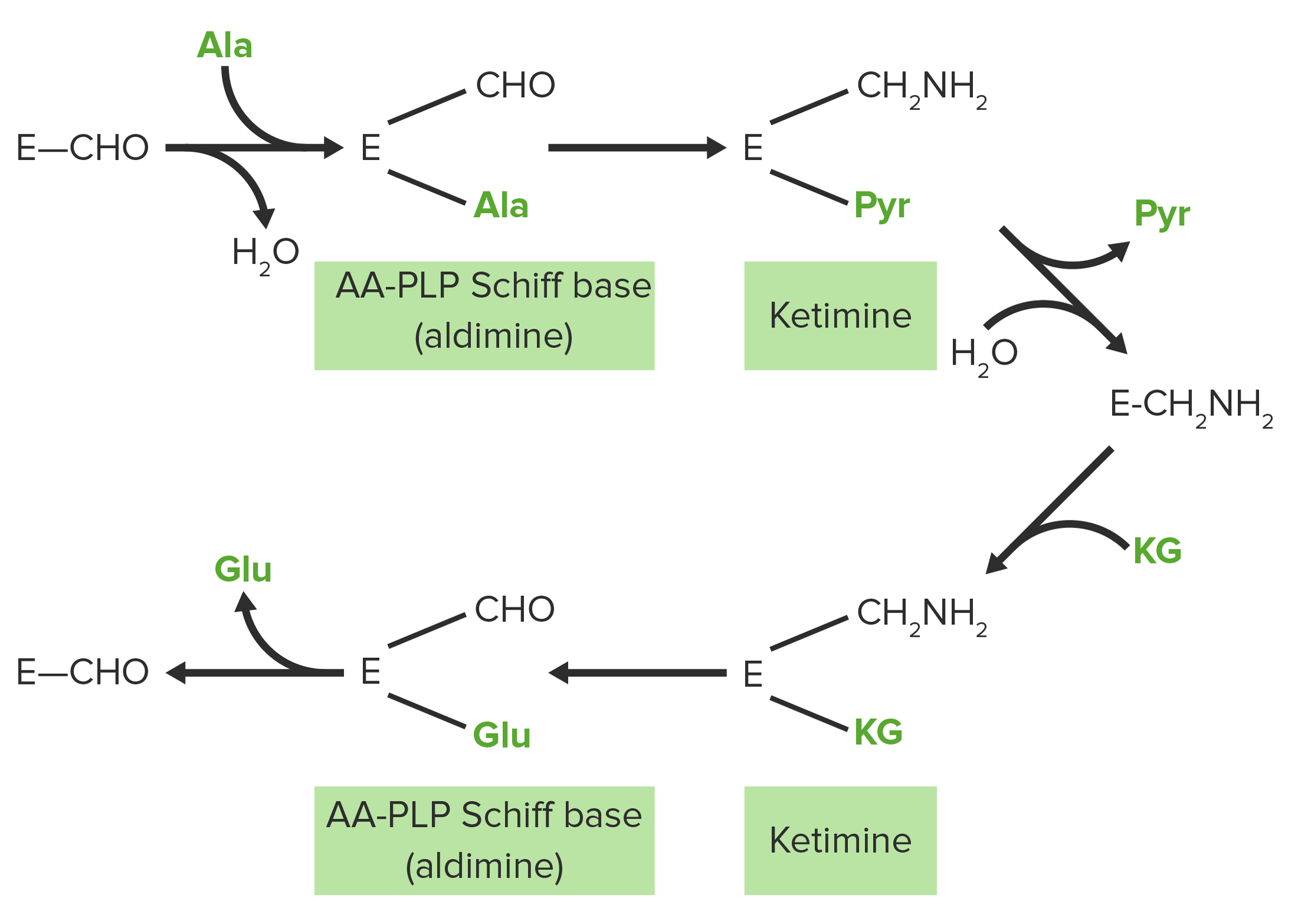
Diagrama esquemático das reações de transaminação de aspartato e glutamato (ácido glutâmico): Os grupos amina estão destacados a vermelho, enquanto os grupos ceto estão destacados a verde.
Imagem por Lecturio
TransaminasesTransaminasesA subclass of enzymes of the transferase class that catalyze the transfer of an amino group from a donor (generally an amino acid) to an acceptor (generally a 2-keto acid). Most of these enzymes are pyridoxyl phosphate proteins.Autoimmune Hepatitis
A alanina transaminaseTransaminaseA subclass of enzymes of the transferase class that catalyze the transfer of an amino group from a donor (generally an amino acid) to an acceptor (generally a 2-keto acid). Most of these enzymes are pyridoxyl phosphate proteins.Catabolism of Amino Acids (ALTALTAn enzyme that catalyzes the conversion of l-alanine and 2-oxoglutarate to pyruvate and l-glutamate.Liver Function Tests ou ALAT) transfere um grupo amina da alanina para o alfa-cetoglutarato, formando piruvato e glutamato.
A aspartato transaminaseTransaminaseA subclass of enzymes of the transferase class that catalyze the transfer of an amino group from a donor (generally an amino acid) to an acceptor (generally a 2-keto acid). Most of these enzymes are pyridoxyl phosphate proteins.Catabolism of Amino Acids (ASTASTEnzymes of the transferase class that catalyze the conversion of l-aspartate and 2-ketoglutarate to oxaloacetate and l-glutamate.Liver Function Tests ou ASAT)transfere um grupo amina de aspartato para alfa-cetoglutarato, formando oxaloacetato e glutamato.
Ambas as enzimas catabolizam reações reversíveis, que são essenciais para o transporte de nitrogénio dos tecidos para o fígado e para o ciclo da ureia.
Mecanismo “bi-bi de pingue-pongue” de transaminação catalisada por enzima dependente de PLP. A reação da aminotransferase ocorre em 2 etapas que consistem em 3 etapas: transaminação, tautomerização e hidrólise. Na primeira etapa, o grupo alfa-amina do aminoácido é transferido para o PLP, produzindo um alfa cetoácido e fosfato de piridoxamina (PMP). Na segunda etapa da reação, o grupo amina de PMP é transferido para um alfa-cetoácido diferente para produzir um novo alfa-aminoácido e PLP.
Imagem por Lecturio.
Etapas :
PLP reage com o grupo amina do AAAAAmyloidosis, libertando H 2 O.
É formada uma base Schiff, desestabilizando o AAAAAmyloidosis.
Os átomos de hidrogénio migram, as ligações duplas mudam e a aldimina → cetimina.
É adicionada H2O, produzindo PMP e um alfa-cetoácido.
Ao contrário, o PMP reage com um alfa-cetoácido, gerando um AAAAAmyloidosis e reconstituindo o PLP.
A desaminação é o processo pelo qual os grupos amino são retirados dos AAs, libertando amónia citotóxica livre: amónia → amónio → ureia ou ácido úrico através do ciclo da ureia no fígado.
Três tipos de desaminação
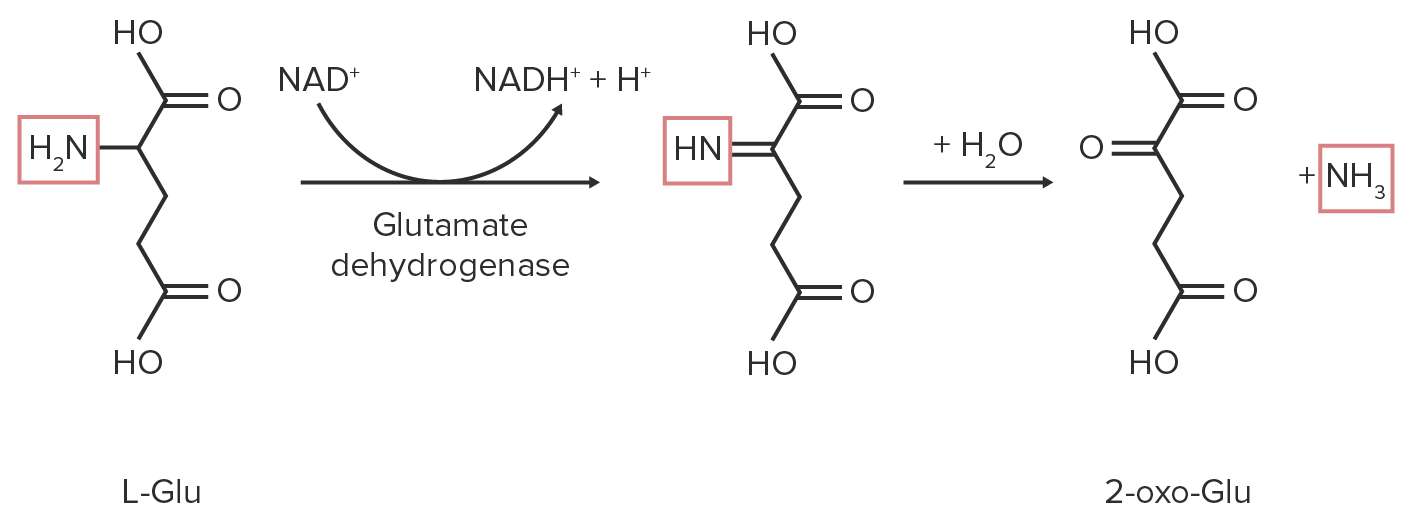
1. Desaminação oxidativa:
A oxidação transforma o grupo amina num grupo imina.
NADNAD+A coenzyme composed of ribosylnicotinamide 5′-diphosphate coupled to adenosine 5′-phosphate by pyrophosphate linkage. It is found widely in nature and is involved in numerous enzymatic reactions in which it serves as an electron carrier by being alternately oxidized (NAD+) and reduced (NADH). Pentose Phosphate Pathway+ ou NADP + é reduzido a NADH/H ou NADPHNADPHNicotinamide adenine dinucleotide phosphate. A coenzyme composed of ribosylnicotinamide 5′-phosphate (nmn) coupled by pyrophosphate linkage to the 5′-phosphate adenosine 2.Pentose Phosphate Pathway/H, respectivamente.
É adicionada água ao grupo amina, convertendo-o em um grupo alfa-ceto, libertando amónia.
Diagrama esquemático da reação de desaminação oxidativa do glutamato. Os grupos funcionais que contêm nitrogénio estão destacados a vermelho.
Imagem por Lecturio.
2. Desaminação hidrolítica:
A água reage com o grupo amina, ligando irreversivelmente um grupo OH e eliminando o grupo amina na forma de amónia.
Diagrama esquemático de uma reação de desaminação hidrolítica. Os grupos funcionais que contêm nitrogénio estão destacados a vermelho.
Imagem por Lecturio.
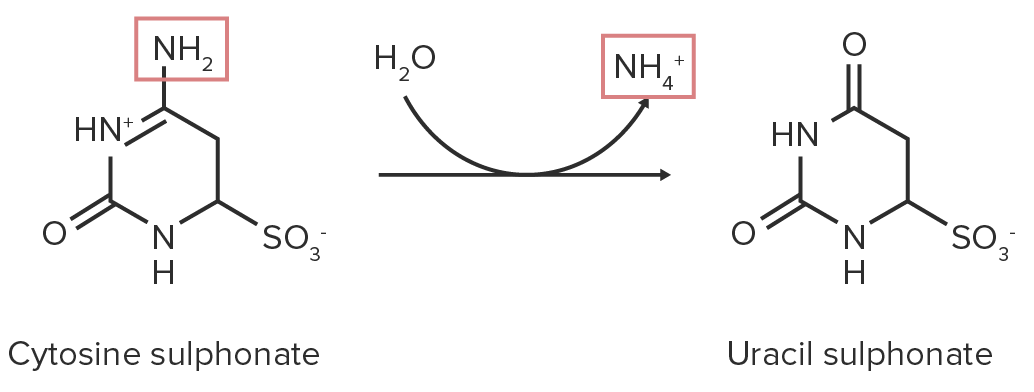
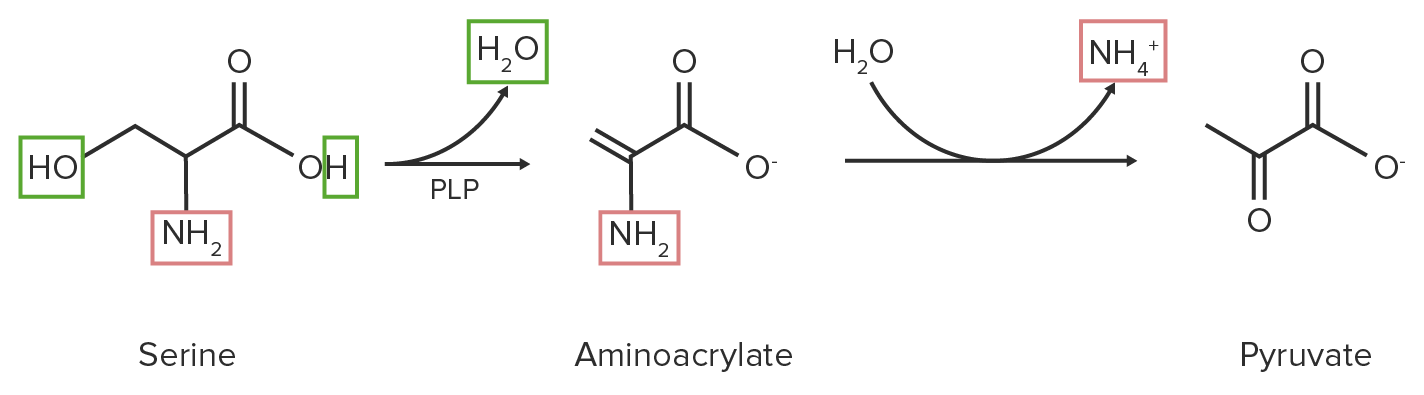
3. Desaminação eliminativa:
Pequenos AAs (serina ou cisteína) libertam água (ou sulfeto de hidrogénio para aminoácidos sulfurosos).
A PLP é uma coenzima necessária.
Através da hidrólise, o grupo amino é clivado, resultando em piruvato.
Diagrama esquemático da reação de desaminação eliminativa da serina. Os grupos funcionais que contêm nitrogénio estão destacados a vermelho, enquanto a molécula de água (H 2 O) e os seus componentes estão destacados a verde.
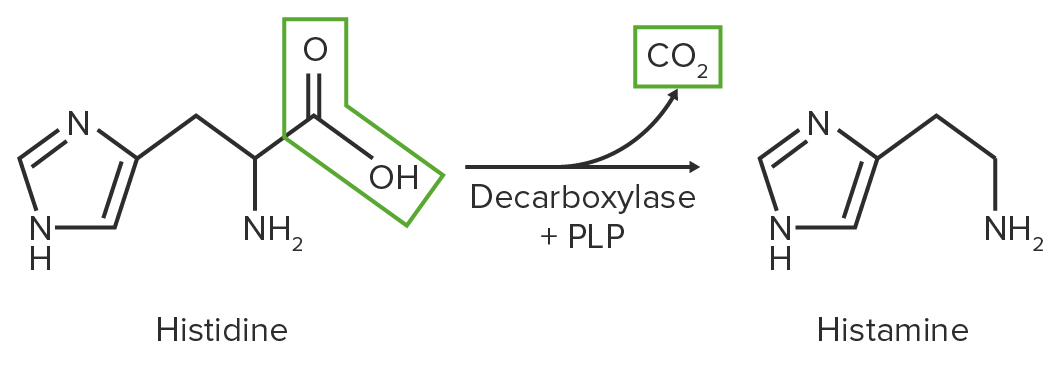
Clivagem de um grupo carboxila de um AAAAAmyloidosis, libertando CO 2
Catalisado pela enzima descarboxilase
Usa PLP como uma coenzima
Aminas resultantes cumprem funções importantes no corpo = aminas biogénicas
A histamina é formada através da descarboxilação da histidina e desempenha um papel vital nas reações de hipersensibilidade imediata.
Outros exemplos:
Acido gama-aminobutírico do ácido glutamina
Dopamina de 3,4-dihidroxifenilalanina
Diagrama esquemático da reação de descarboxilação de histidina em histamina
Imagem por Lecturio.
Catabolismo do Esqueleto de Carbono
O catabolismo dos AAs envolve reações anapleróticas (reações químicas que formam intermediários das vias metabólicas).
A degradação do esqueleto de carbono dos AAs pode ser classificada pelas vias metabólicas para as quais os seus produtos catabólicos servem como intermediários:
AAs glicogénicos → intermediários da gliconeogénese
AAs cetogénicos → intermediários da cetogénese
AAs glicogénicos e cetogénicos → ambas as vias
AAs glicogénicos
AAs Cetogênicos
AAs Glicogénicos/Cetogénicos
Alanina
Arginina
Asparagina
Ácido aspártico
Cisteína
Ácido glutâmico
Glutamina
Glicina
Histidina
Metionina
Prolina
Serina
Valina
Lisina
Leucina
Isoleucina
Fenilalanina
Treonina
Triptofano
Tirosina
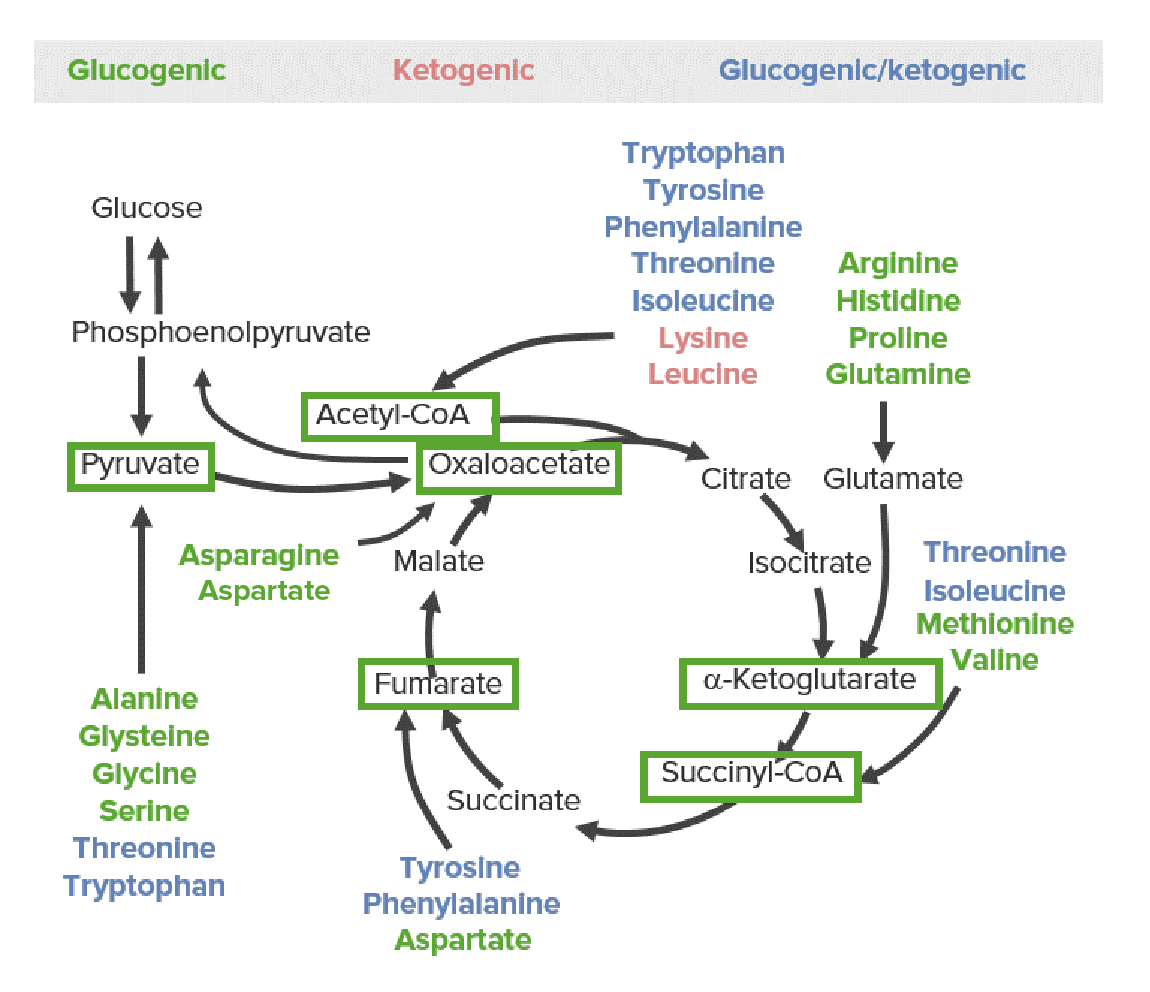
Todos os AAs são divididos em 1 de 6 intermediários (ver as caixas verdes nas imagens abaixo): piruvato, acetil-CoA, oxaloacetato, alfa-cetoglutarato, succinil-CoA ou fumarato.
As 3 categorias de produtos catabólicos de aminoácidos: glicogénicos (verde), cetogénicos (vermelho) e glicogénicos e cetogénicos (azul). A via glicose-piruvato à esquerda representa a glicólise e a gliconeogénese. A via cíclica à direita representa o ciclo do ácido cítrico. Todos os aminoácidos são divididos em 1 de 6 intermediários (caixas verdes): piruvato, acetil-CoA, oxaloacetato, alfa-cetoglutarato, succinil-CoA ou fumarato.
AAs glicogénicos
Metabolizados em piruvato ou metabolitos do ciclo do ácido cítrico (CAC):
Piruvato (de serina, cisteína, glicina, alanina e treonina)
Succinil-CoA (de metionina, isoleucina, valina e treonina via propionil-Coa e metilmalonil-CoA
Propionil-CoA (intermediário da via succinil-CoA)
Oxaloacetato (da asparagina via aspartato)
α-cetoglutarato (de glutamina, arginina, histidina, prolina via glutamato)
Fumarato
Os produtos catabólicos movem-se para o CAC para produzir energia ou são usados como substratos para a gliconeogénese.
AAs cetogénicos
Metabolizados diretamente em acetil-CoA, então entra em 1 de 3 vias metabólicas:
Entram no CAC para produzir ATP/energia
Cetogénese (produção de corpos cetónicos)
Síntese de ácidos gordos ou colesterol
AAs glicogénicos e cetogénicos
Metabolizados em intermediários das vias lipídicas e glicogénicas:
Isoleucina → propionil-CoA (→ metilmalonil-CoA → succinil-CoA) e acetil-CoA
Fenilalanina → tirosina → fumarato e acetil-CoA
Treonina → propionil-CoA e piruvato, bem como acetil-CoA (via glicina + acetaldeído)
Triptofano → alanina e acetil-CoA
Mnemónica
Para relembrar as vias metabólicas dos esqueletos de carbono dos aminoácidos, lembrar:
Cetogénico: “The onLy pureLy ketogenic aminoacids.”
Leucina
Lisina
Relevância Clínica
As seguintes condições são doenças do metabolismo de aminoácidos . Dependendo do país e do estado dos EUA, os recém-nascidos podem ser examinados rotineiramente para estas doenças (exceto para alcaptonúria).
Fenilcetonúria: defeito da fenilalanina hidroxilase que resulta no comprometimento da conversão de fenilalanina em tirosina e subsequente acumulação de fenilalanina. Apresenta-se como atraso psicomotor e convulsões
“Maple syrup urine disease”: defeito na desidrogenase que resulta na acumulação de AAs de cadeia ramificada. Apresenta-se como deficiência cognitiva, urina com cheiro adocicado e distonia
Homocistinúria: defeito na enzima cistationina β-sintase, que leva à acumulação de homocisteína. Apresenta-se como ruborRuborInflammation, atraso no desenvolvimento, luxação do cristalino, doença vascular e osteoporose
Tirosinemia: deficiência de fumarilacetoacetato hidrolase, a última enzima do catabolismo da tirosina. Apresenta-se como doença hepática, ganho de peso insuficiente, doença dos nervos periféricos e defeitos renais
Alcaptonúria: deficiência da dioxigenase do ácido homogentísico, que afeta a degradação normal da tirosina em fumarato. Apresenta-se como uma descoloração preto-azulada dos tecidos conjuntivos, artrite e calcificações de vários tecidos.
Guoyao, W. (2022). Amino acids: Biochemistry and nutrition (2nd ed.). CRC Press.
Jiang, H., Wang, Y., & Li, M. (2025). Role of branched-chain amino acid catabolism in the regulation of adipocyte metabolism. Endocrinology, bqaf089. https://doi.org/10.1210/endocr/bqaf089
Knol, M. G. E., Verhave, J. C., & Navis, G. (2024). Amino acid metabolism in kidney health and disease. Nature Reviews Nephrology, 20(12), 771–788. https://doi.org/10.1038/s41581-024-00872-8
Ling, Z.-N., Jiang, Y.-F., Ru, J.-N., Lu, J.-H., & Ding, B. (2023). Amino acid metabolism in health and disease. Signal Transduction and Targeted Therapy, 8, 345. https://doi.org/10.1038/s41392-023-01569-3
Nelson, D. L., & Cox, M. M. (2021). Lehninger principles of biochemistry (8th ed.). W. H. Freeman and Company.
Rodwell, V. W., Bender, D. A., Botham, K. M., Kennelly, P. J., & Weil, P. A. (2022). Harper’s illustrated biochemistry (32nd ed.). McGraw-Hill Education.
Torres, N., Tovar, A. R., & Torres, L. F. (2023). Amino acid catabolism: An overlooked area of metabolism. Nutrients, 15(15), 3378. https://doi.org/10.3390/nu15153378
Crie sua conta gratuita ou faça login para continuar lendo!
A Lecturio Medical complementa o teu estudo através de métodos de ensino baseados em evidência, vídeos de palestras, perguntas e muito mais – tudo combinado num só lugar e fácil de usar.
User Reviews
Details
×
Obtenha Premium para testar os seus conhecimentos
Lecturio Premium dá-lhe acesso total a todos os conteúdos e características
Obtenha Premium para ver todos os vídeos
Verifique agora o seu e-mail para obter um teste gratuito.
Crie uma conta gratuita para testar os seus conhecimentos
Lecturio Premium dá-lhe acesso total a todos os conteúdos e características - incluindo o Qbank de Lecturio com perguntas actualizadas ao estilo do board-.