


A síndrome de insensibilidade a androgénios (SIA) é uma doença recessiva ligada ao X em que uma mutação genética afeta a função dos recetores de androgénios, resultando em resistência completa (SIAC), parcial (SIAP) ou ligeira (SIAL) à testosterona. Todos os indivíduos com SIA têm cariótipo 46,XY; no entanto, os fenótipos variam e incluem indivíduos com fenótipo feminino, feminino virilizado, masculino não virilizado e indivíduos com fenótipo masculino. A síndrome de insensibilidade aos androgénios conduz sempre a infertilidade. As análises hormonais, os exames de imagem e os testes Testes Gonadal Hormones genéticos ajudam a fazer o diagnóstico. O tratamento da SIA varia de acordo com o grau de sensibilidade aos androgénios, fenótipo e identidade de género, e pode envolver terapia de reposição hormonal e cirurgia para corrigir anomalias anatómicas das estruturas genitais e reprodutivas.
Last updated: Dec 15, 2025
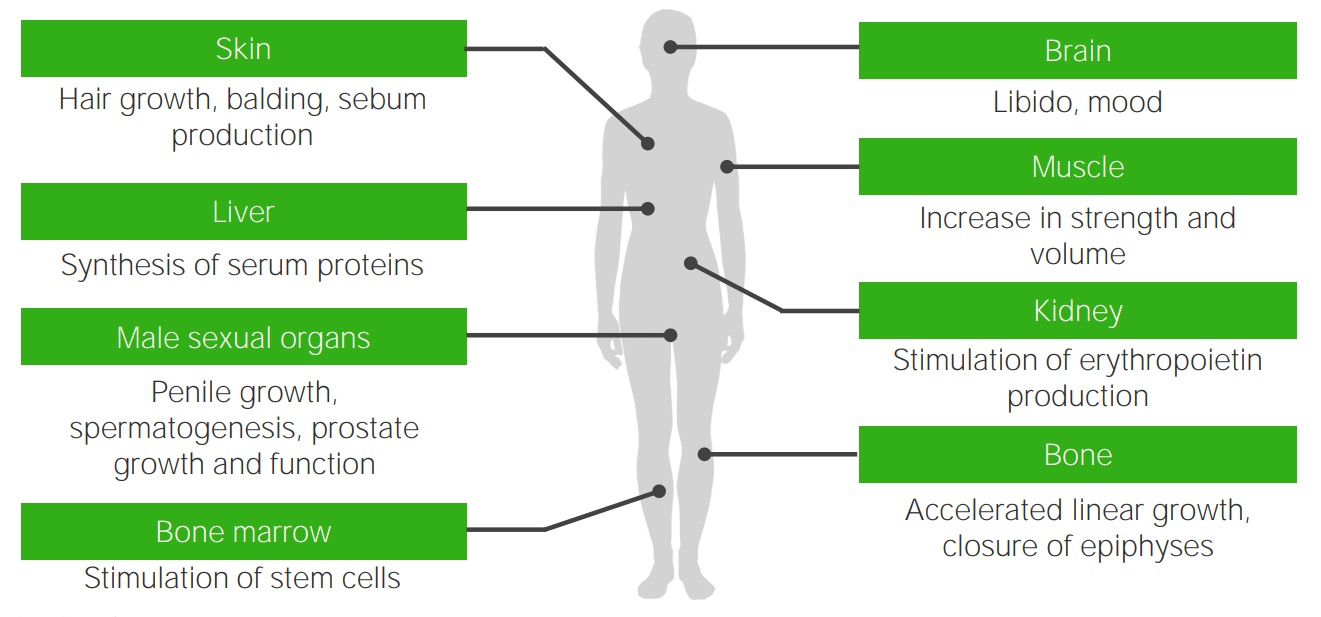
Efeitos da testosterona: órgãos-alvo da testosterona e os efeitos androgénicos típicos ausentes ou deficientes nos indivíduos com síndrome de insensibilidade aos androgénios
Imagem por Lecturio.As características sexuais dos indivíduos afetados podem variar e levar a 1 dos 3 tipos de classificação:
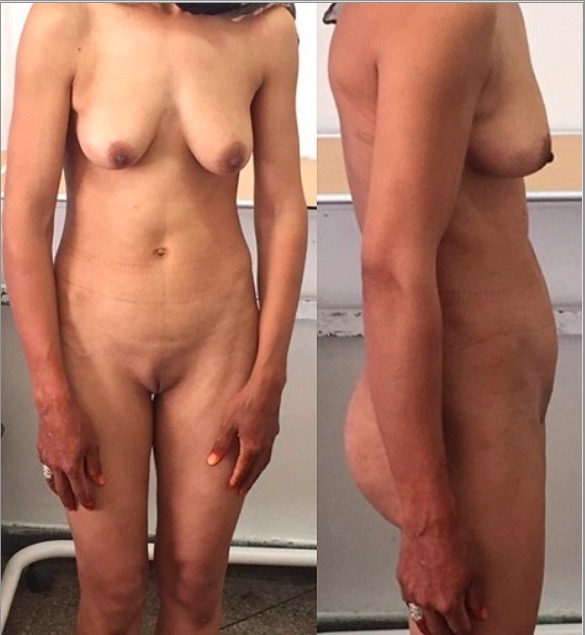
Síndrome de insensibilidade aos androgénios completa: uma paciente de 30 anos que apresenta amenorreia primária. Observe os pelos púbicos escassos.
Imagem: “Front and side view of the patient” por Regragui Souhail et al. Licença: CC BY 2.0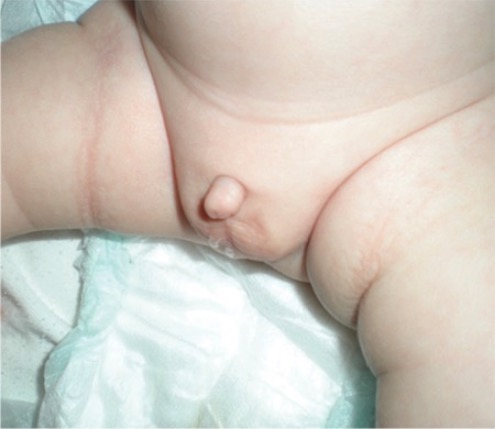
Criança nascida com micropénis
Imagem: “Micropenis in a newborn” por Erciyes University Faculty of Medicine, Department of Pediatric Endocrinology, Kayseri, Turkey. Licença: CC BY 2.5
Síndrome de insensibilidade parcial aos androgénios (SIAP)
Um paciente com SIAP que se apresentou com um fenótipo masculino com ginecomastia
Deve considerar-se todas as perturbações que afetam o desenvolvimento sexual. A seguir estão algumas doenças selecionadas que fazem parte dos diagnósticos diferenciais da SIA:
A doença seguinte está relacionada com a SIA, porque envolve o gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics RA e compromete a recetividade dos androgénios:
Atrofia muscular espinobulbar: uma doença genética ligada ao X, também conhecida como doença de Kennedy. A atrofia muscular espinobulbar é causada pela repetição da expansão anormal no gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics RA, resultando em compromisso do RA. A atrofia muscular espinobulbar é caracterizada por uma deterioração progressiva dos neurónios motores anteriores e está associada ao desenvolvimento de ginecomastia, infertilidade e perfis hormonais semelhantes aos observados na SIA.
