Los LOS Neisseria trastornos del almacenamiento de glucógeno son defectos genéticos que conducen a trastornos del metabolismo de los LOS Neisseria carbohidratos. Los LOS Neisseria trastornos son causados por variantes patogénicas en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure que afectan a las enzimas involucradas en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la degradación del glucógeno. La deficiencia de 1 de estas enzimas puede ocurrir en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el hígado o los LOS Neisseria músculos y puede causar hipoglucemia y/o depósito anormal de glucógeno en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria tejidos. Las presentaciones varían desde ser fatal en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el periodo neonatal hasta tener una presentación inicial de los LOS Neisseria síntomas en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la adultez. Hay al AL Amyloidosis menos 14 tipos de trastornos del almacenamiento de glucógeno, y los LOS Neisseria 4 más comunes y significativos son la enfermedad de von Gierke, la enfermedad de Pompe, la enfermedad de Cori y la enfermedad de McArdle. El diagnóstico es clínico; la detección de glucógeno en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria tejidos se realiza mediante biopsia y se confirma mediante análisis del ácido desoxirribonucleico (ADN). El tratamiento tiene como objetivo tratar o evitar la hipoglucemia, hiperuricemia, hiperlipidemia y acidosis Acidosis A pathologic condition of acid accumulation or depletion of base in the body. The two main types are respiratory acidosis and metabolic acidosis, due to metabolic acid build up. Respiratory Acidosis láctica. Actualmente, no hay cura disponible, pero se están probando terapias genéticas.
Last updated: Apr 17, 2025
Los LOS Neisseria trastornos del almacenamiento de glucógeno son defectos genéticos que causan deficiencias enzimáticas que resultan en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum enfermedades del hígado, músculo o corazón, desde depósitos anormales de glucógeno en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria tejidos y episodios de hipoglucemia ( en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la mayoría de las enfermedades), ya que el cuerpo no puede usar el glucógeno como fuente de energía.
El trastorno por almacenamiento de glucógeno I (enfermedad de von Gierke) es causado por mutaciones que conducen a deficiencias enzimáticas; estos dan como resultado un exceso de glucógeno y acumulación de grasa en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria tejidos e hipoglucemia.
El trastorno de almacenamiento de glucógeno II (enfermedad de Pompe) es un trastorno de almacenamiento lisosomal autosómico recesivo causado por una variante patogénica en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum GAA. El defecto da como resultado una deficiencia de la enzima alfa-glucosidasa, con la consiguiente acumulación de glucógeno en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el músculo cardíaco y esquelético.
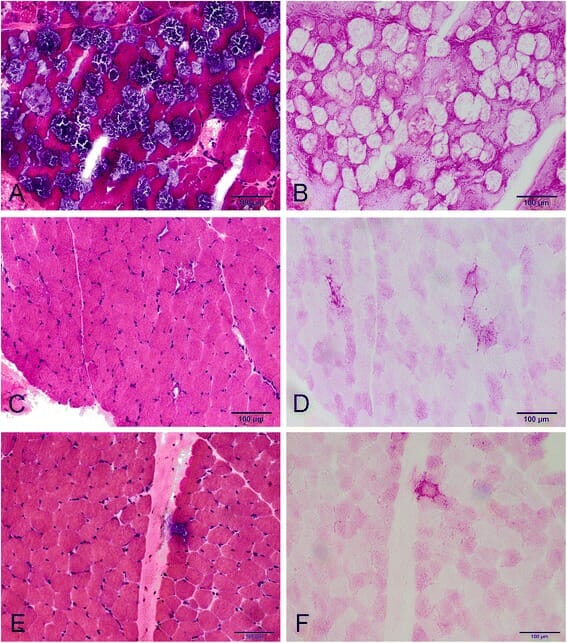
Cambios miopáticos con la enfermedad de Pompe de inicio tardío:
A: tinción de hematoxilina-eosina (H&E)—vacuolización extensa en muchas fibras.
C y E: tinción H&E—vacuolación en solo unas pocas fibras en un paciente diferente.
B, D y F: tinción de ácido peryódico de Schiff (PAS, por sus siglas en inglés): fibras vacuolares teñidas positivas para glucógeno.
Características clínicas de la enfermedad de Pompe:
Atrofia del músculo cuádriceps (A), alas escapulares (B) y ptosis (C) como características clínicas notables en adultos con enfermedad de Pompe. Las fotografías se imprimen con el permiso de los pacientes.
El trastorno de almacenamiento de glucógeno III (enfermedad de Cori) es causado por una deficiencia en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum las enzimas desramificadoras de glucógeno y conduce a la deposición de glucógeno en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el hígado, músculos y corazón.
La enfermedad de almacenamiento de glucógeno V (enfermedad de McArdle), también conocida como deficiencia de miofosforilasa, es un trastorno autosómico recesivo causado por mutaciones en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la isoforma muscular de la fosforilasa (glucógeno fosforilasa muscular o PYGM) ubicada en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum 11q13.
| von Gierke (trastorno de almacenamiento de glucógeno I) | Pompe (trastorno de almacenamiento de glucógeno II) | Cori (trastorno de almacenamiento de glucógeno III) | McArdle (trastorno de almacenamiento de glucógeno V) | |
|---|---|---|---|---|
| Presentación |
|
|
|
|
| Diagnóstico |
|
|
|
|
| Tratamiento |
|
|
|
|