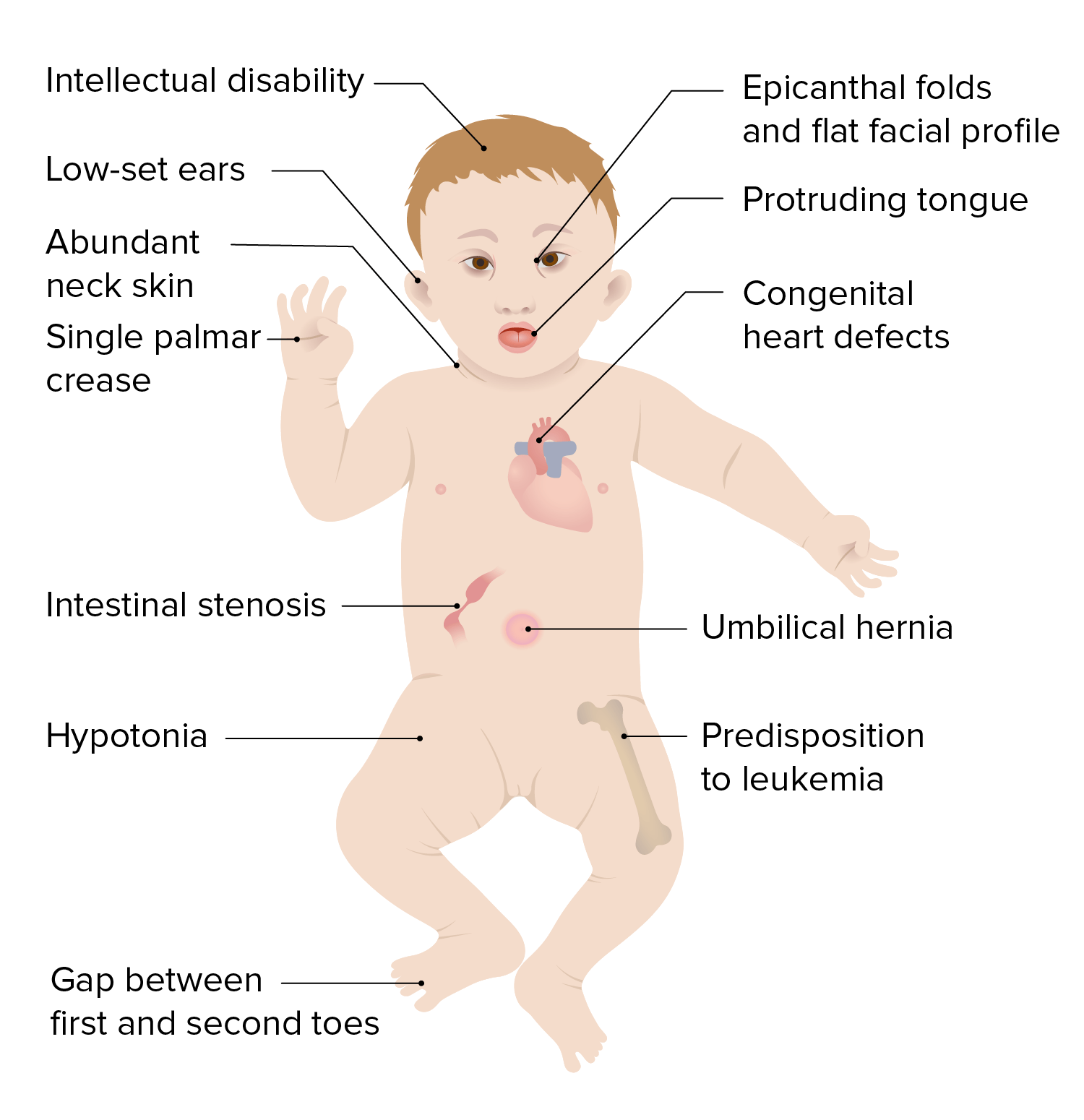
El síndrome de Down, o trisomía 21, es la aberración cromosómica más común y la causa genética más frecuente de retraso en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el desarrollo. Tanto los LOS Neisseria niños como las niñas están afectados y presentan rasgos craneofaciales y musculoesqueléticos característicos, así como múltiples anomalías médicas que afectan a los LOS Neisseria sistemas cardíaco, gastrointestinal, ocular y auditivo. Los LOS Neisseria rasgos característicos son los LOS Neisseria ojos inclinados hacia arriba, en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum forma de almendra, con la piel cubriendo los LOS Neisseria epicantos internos, el puente nasal ancho y aplanado, las orejas pequeñas y redondeadas y la boca pequeña con una lengua grande. El tamizaje para síndrome de Down se realiza durante el primer y segundo trimestre del embarazo e incluye tanto análisis de sangre como ultrasonidos prenatales. En EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la última instancia, el cariotipo confirma el diagnóstico en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el período prenatal o postnatal. El síndrome de Down no tiene cura. El tratamiento se basa en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum las manifestaciones clínicas presentes; incluye un sólido sistema de apoyo y programas de intervención temprana para ayudar a la educación y el desarrollo.
Last updated: Mar 19, 2025
Existen 3 genotipos que dan lugar al AL Amyloidosis síndrome de Down.
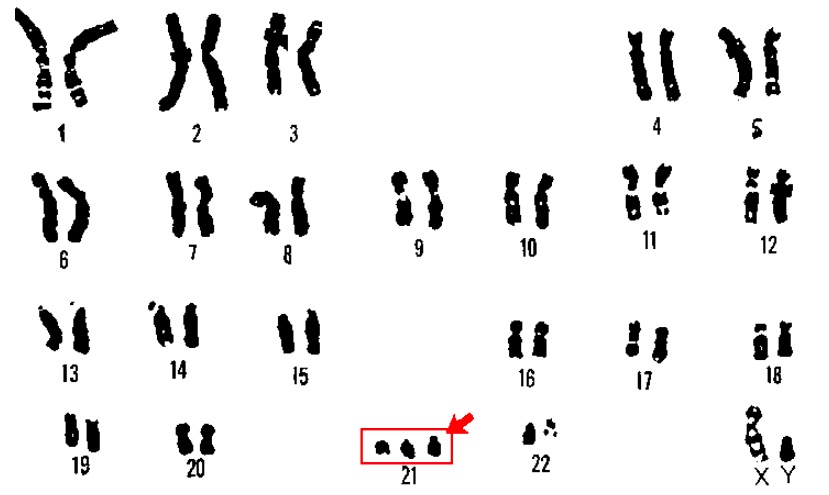
Cariotipo de la trisomía 21 (síndrome de Down):
El cariotipo es una técnica de laboratorio en la que se emparejan y fotografían los cromosomas de un individuo. En un paciente con síndrome de Down suelen ser visibles 3 copias del cromosoma 21.
Esquema de una mutación genética, en este caso de la trisomía 21
Imagen: “Genome scheme of trisomy 21” por National Human Genome Research Institute. Licencia: Dominio Público
Translocación del síndrome de Down
Imagen: “Down syndrome translocation” por National Human Genome Research Institute. Licencia: Dominio Público
Translocación robertsoniana del síndrome de Down en familias:
En una translocación robertsoniana causante del síndrome de Down, parte de un cromosoma 21 extra o uno completo se une (transloca) a otro cromosoma. Los individuos que heredan este cromosoma tienen 3 copias del material genético que se encuentra en el cromosoma 21, por lo que tienen síndrome de Down.

Los ojos de un bebé con síndrome de Down:
Las manchas de Brushfield son visibles entre los círculos internos y externos del iris.
Alta prevalencia de trastornos oculares: La frecuencia aumenta con la edad.
Pies de un niño de 10 años con síndrome de Down, con el típico espacio grande entre el dedo gordo y el segundo dedo
Imagen: “Feet of a boy with Down syndrome” por Loranchet. Licencia: CC BY 3.0Se recomienda el tamizaje antes de la semana 20 de gestación.
Trisomía 21 en el ultrasonido:
Imagen ecográfica del feto a las 12 semanas, que muestra la translucidez nucal y la presencia de hueso nasal
Dada la naturaleza omnipresente del síndrome de Down y la falta de cura, la mayor parte del tratamiento se centra en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el tamizaje y la prevención de las diversas manifestaciones clínicas y complicaciones asociadas al AL Amyloidosis síndrome.