La polimiositis es una miopatía inflamatoria autoinmune causada por una lesión muscular mediada por linfocitos T. La etiología de la polimiositis no está clara, pero existen varias asociaciones genéticas y ambientales. La polimiositis es más común en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum mujeres de mediana edad y rara vez afecta a los LOS Neisseria niños. Los LOS Neisseria pacientes presentan debilidad muscular proximal progresiva y simétrica y síntomas constitucionales. Las complicaciones pueden surgir de la afectación respiratoria, cardíaca o gastrointestinal. El diagnóstico se basa en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la presentación clínica y estudios de laboratorio y se confirma mediante una biopsia muscular. El tratamiento es con corticosteroides sistémicos, inmunosupresores y fisioterapia. Todos los LOS Neisseria pacientes deben someterse a exámenes de tamizaje de cáncer porque existe una fuerte asociación con la malignidad.
Last updated: Dec 15, 2025
La polimiositis es una miopatía inflamatoria autoinmune que se presenta como debilidad muscular proximal simétrica. El síndrome antisintetasa es un subtipo que muestra la presencia de anticuerpos antisintetasa y ciertas manifestaciones extramusculares.
Se desconoce la etiología exacta.
Posible predisposición genética:
Factores ambientales:
Afecciones médicas asociadas:
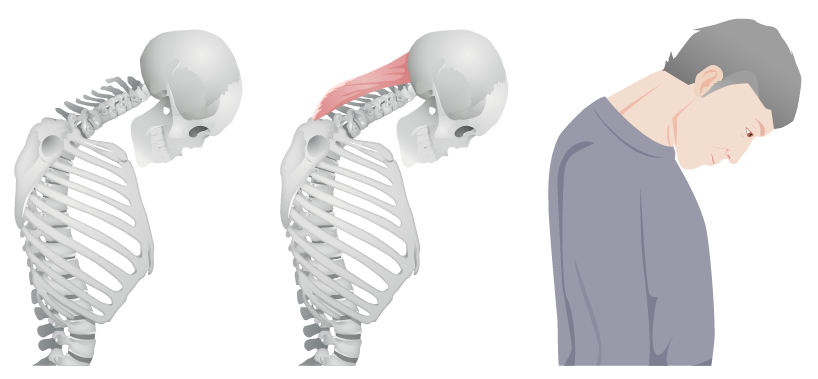
“Cabeza caída” en polimiositis: causada por debilidad en los músculos extensores del cuello
Imagen por Lecturio.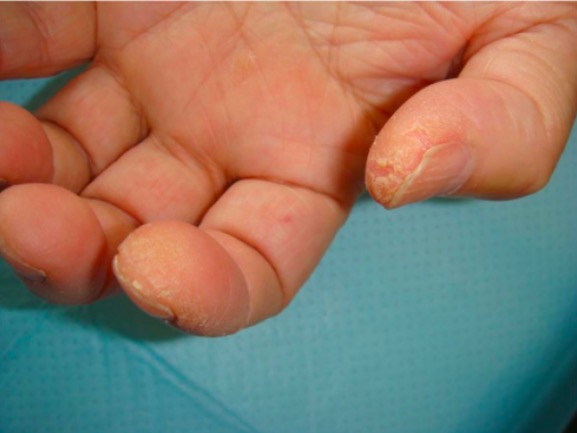
“Manos de mecánico” asociadas a anticuerpos antisintetasa en la polimiositis: hiperqueratosis que produce fisuras, rugosidad y descamación en la yema del pulgar y la cara lateral del dedo índice
Imagen: “Patient 3” por Department of Respiratory Medicine, Kyorin University School of Medicine, 6-20-2 Shinkawa, Mitaka City, Tokyo 181-8611, Licencia: CC BY 2.0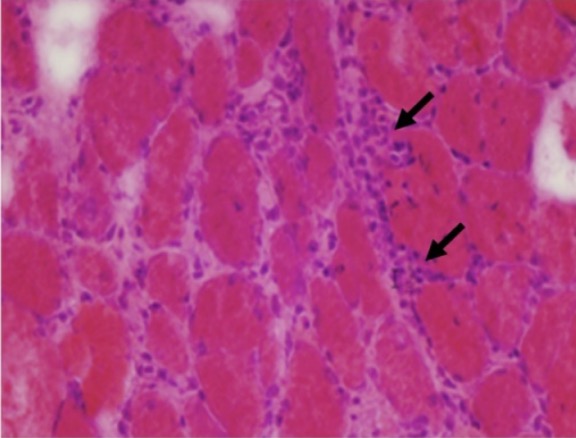
Hallazgos histológicos en la polimiositis:
Biopsia muscular de un paciente con polimiositis que muestra células inflamatorias del endomisio (flechas) y variaciones en el tamaño de las fibras

Hallazgos electromiográficos en la polimiositis: gráfico comparativo que muestra las diferencias en la actividad electromiográfica para diferentes patologías
Imagen por Lecturio.Se utiliza un abordaje multidisciplinario que incluye terapias farmacológicas y no farmacológicas en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el tratamiento de la polimiositis.