LosLOSNeisseriahemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease son neoplasias vasculares del SNC. LosLOSNeisseriahemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease son raros y a menudo se asocian con la enfermedad de von Hippel-Lindau. La presentación más común es cefalea y, según el tamaño y la ubicación del tumorTumorInflammation, losLOSNeisseria pacientes pueden presentar déficits sensoriales y debilidad motora. La imagenología es el principal método diagnóstico y se requiere una evaluación histopatológica para el diagnóstico definitivo. La cirugía a menudo está indicada enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el tratamiento de losLOSNeisseriahemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease, aunque según el tamaño, el número y la ubicación de losLOSNeisseria tumores, también se puede justificar la radioterapia. El pronóstico suele ser bueno enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseriahemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease solitarios, pero losLOSNeisseria tumores asociados con von Hippel-Lindau a menudo se asocian con un peor pronóstico y un mayor riesgo de recurrencia.
LosLOSNeisseriahemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Diseaseson neoplasias del SNC raras, de crecimiento lento, benignas y altamente vascularizadas que tienen una alta asociación con la enfermedad de von Hippel-Lindau, que es una condición autosómica dominante caracterizada por una variedad de tumores benignos y malignos.
LosLOSNeisseria tumores se encuentran con mayor frecuencia enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el cerebelo.
LosLOSNeisseriahemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease suelen encontrarse enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el parénquima unido a la piamadre o cerca de ella, una parte de las meningesMeningesThe brain and the spinal cord are enveloped by 3 overlapping layers of connective tissue called the meninges. The layers are, from the most external layer to the most internal layer, the dura mater, arachnoid mater, and pia mater. Between these layers are 3 potential spaces called the epidural, subdural, and subarachnoid spaces. Meninges: Anatomy (formadas por 3 capas fibrosas) que recubren el SNC:
Duramadre: la capa externa, que forma una cubierta de colágeno resistente
Aracnoides: capa intermedia que consta de epitelio escamoso simple con una malla suelta de colágeno y fibras elásticas
Piamadre: capa interna delgada enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum contacto con el cerebro y la médula espinal
Tabla: Clasificación de losLOSNeisseria tumores del sistema nervioso
Categorías
Tumores específicos
Tumores neuroepiteliales enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el SNC
Astrocitomas, incluido el glioblastoma multiformeGlioblastoma multiformeGlioblastoma multiforme is a high-grade astrocytoma, an aggressive brain tumor arising from astrocytes, with an unknown cause and a poorly understood link to risk factors. There are two main types: primary, a more aggressive form seen more commonly in older patients, and secondary, developing from lower-grade astrocytomas and seen more commonly in younger patients.Glioblastoma Multiforme
OligodendrogliomaOligodendrogliomaOligodendrogliomas are malignant CNS tumors arising from neural glial cell precursors. Oligodendrogliomas often arise in the frontal lobes of the brain and have a generally favorable prognosis when compared to other gliomas. Oligodendrogliomas are the 3rd most common CNS tumor. The most frequent presenting symptom is a seizure.Oligodendroglioma
Ependimoma y tumores del plexo coroideo
Meduloblastomas (tumores embrionarios)
Tumores meníngeos
Meningiomas
HemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease
Tumores de la región selar
Craneofaringioma
Adenoma hipofisario
Pinealoma/pinealoblastoma
Linfoma primario del SNC
Linfoma primario del SNC
Metástasis alALAmyloidosis cerebro (5 veces más común que losLOSNeisseria tumores cerebrales primarios)
Más comúnmente surgen de:
Carcinomas de pulmón, mama y células renales
MelanomaMelanomaMelanoma is a malignant tumor arising from melanocytes, the melanin-producing cells of the epidermis. These tumors are most common in fair-skinned individuals with a history of excessive sun exposure and sunburns. Melanoma
NeuroblastomaNeuroblastomaNeuroblastoma is a malignancy that arises from the neural crest cell derivatives along the sympathetic chain (neuroblasts) and is most commonly located in the adrenal medulla. The tumor often presents in childhood with a flank mass that crosses the midline.Neuroblastoma
Epidemiología
Esporádico: 75%
Asociado a von Hippel-Lindau: 25%
Incidencia:
Extremadamente raro
2,5% de todas las neoplasias intracraneales
5% de losLOSNeisseria tumores de la médula espinal
Sexo: La incidencia es mayor enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum hombres que enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum mujeres (2:1).
Se desconoce la causa exacta del hemangioblastomaHemangioblastomaHemangioblastomas are vascular neoplasms of the CNS. Hemangioblastomas are rare and are often associated with von Hippel-Lindau disease (VHL). The most common presentation is a headache and, depending on the size and location of the tumor, patients may present with sensory deficits and motor weakness.Hemangioblastoma.
Se está considerando una mutación enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el gen VHL debido a una alta asociación con von Hippel-Lindau.
LosLOSNeisseriahemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease crecen adheridos a la piamadre (capa meníngea más interna)
Parénquima del cerebelo
Tronco encefálico
Médula espinal
Patogénesis
Aunque losLOSNeisseria hemangiomas esporádicos no tienen una patogénesis clara, se cree que losLOSNeisseria hemangiomas asociados con von Hippel-Lindau son causados por una mutación enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el gen VHL. Hasta el 50% de losLOSNeisseriahemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease esporádicos también tienen mutaciones o deleciones enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el gen VHL.
Gen VHL:
Gen supresor de tumores encontrado enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el cromosoma 3p
Responsable de la inhibición del factor 2α inducible por hipoxia (HIF-2α) por degradación proteasomal mediada por ubiquitina
HIF-2α:
Parte de un complejo de proteína de factor de transcripción más grande llamado HIFHIFHypoxia-inducible factor 1, alpha subunit is a basic helix-loop-helix transcription factor that is regulated by oxygen availability and is targeted for degradation by VHL tumor suppressor protein.Von Hippel-Lindau Disease, que participa enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la regulación de la capacidad del cuerpo para adaptarse a losLOSNeisseria cambios enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria niveles de oxígeno.
HIFHIFHypoxia-inducible factor 1, alpha subunit is a basic helix-loop-helix transcription factor that is regulated by oxygen availability and is targeted for degradation by VHL tumor suppressor protein.Von Hippel-Lindau Disease puede inducir la expresión de más de 70 genesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure diana, que incluyen:
Factor de crecimiento endotelial vascular (VEGF, por sus siglas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum inglés)
Factor de crecimiento derivado de plaquetas (PDGF, por sus siglas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum inglés)
Eritropoyetina
Factor de crecimiento transformante α (TGF-α)
Cuando hay suficiente oxígeno disponible, la proteína VHL ayuda a suprimir el HIFHIFHypoxia-inducible factor 1, alpha subunit is a basic helix-loop-helix transcription factor that is regulated by oxygen availability and is targeted for degradation by VHL tumor suppressor protein.Von Hippel-Lindau Disease.
Un gen VHL disfuncional (causado por mutación o deleción) puede conducir a la acumulación de HIF-α debido a la incapacidad de degradar HIF-2α.
La estabilización de HIF-2α le permite inducir la expresión de sus genesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure diana → ↑ factores angiogénicos → crecimiento tumoral
Desarrollo de síntomas
El desarrollo de losLOSNeisseria síntomas es causado por:
Compresión directa del tumorTumorInflammation sobre las estructuras neurales
DolorDolorInflammation local (común enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum lesiones de la médula espinal)
AtaxiaAtaxiaImpairment of the ability to perform smoothly coordinated voluntary movements. This condition may affect the limbs, trunk, eyes, pharynx, larynx, and other structures. Ataxia may result from impaired sensory or motor function. Sensory ataxia may result from posterior column injury or peripheral nerve diseases. Motor ataxia may be associated with cerebellar diseases; cerebral cortex diseases; thalamic diseases; basal ganglia diseases; injury to the red nucleus; and other conditions.Ataxia-telangiectasia cerebelosa y descoordinación
Aumento de la presión intracraneal (PIC) debido a hidrocefalia obstructiva
Disfunción del nervio oculomotor
Debilidad motora
Déficits sensoriales
HemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease esporádicos versus asociados a von Hippel-Lindau
Tabla: Presentaciones clínicas típicas de hemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease esporádicos versus asociados a Von Hippel-Lindau
HemangioblastomaHemangioblastomaHemangioblastomas are vascular neoplasms of the CNS. Hemangioblastomas are rare and are often associated with von Hippel-Lindau disease (VHL). The most common presentation is a headache and, depending on the size and location of the tumor, patients may present with sensory deficits and motor weakness.Hemangioblastoma esporádico
HemangioblastomaHemangioblastomaHemangioblastomas are vascular neoplasms of the CNS. Hemangioblastomas are rare and are often associated with von Hippel-Lindau disease (VHL). The most common presentation is a headache and, depending on the size and location of the tumor, patients may present with sensory deficits and motor weakness.Hemangioblastoma asociado a von Hippel-Lindau
Generalmente ocurre enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la 3ra o 4ta década de la vida
Diagnosticado a una edad más temprana (2da década)
Comúnmente se presenta como un tumorTumorInflammation aislado enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el cerebelo
El 50% de losLOSNeisseria tumores se localizan enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la médula espinal, el 40% enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el cerebelo y el 10% enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el tronco encefálico.
Complicaciones
Las complicaciones suelen ocurrir por aumento del tamaño del tumorTumorInflammation (> 1,5 cm), provocando compresión, o por hemorragia espontánea:
Síntomas de aumento de la PIC/hidrocefalia obstructiva rápida
Estado mental alterado
Policitemia por producción ectópica de eritropoyetina (síndrome paraneoplásico)
Cuadriplejia
Diagnóstico
Si bien la principal herramienta diagnóstica es la imagenología, el estándar de oro del diagnóstico requiere la histopatología de una muestra de biopsia.
Imagenología
Se deben obtener imágenes de todo el eje neural para descartar lesiones múltiples, que son comunes enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria casos de von Hippel-Lindau.
RM de contraste:
La RM realzada con gadolinio es el método de imagenología más sensible para diagnosticar el hemangioblastoma.
Rasgos característicos:
Nódulo realzado asociado a un quiste (60% de los tumores)
Tumor sólido realzado (40%)
T1 muestra un nódulo hipointenso a isointenso
T2 muestra un nódulo hiperintenso
Cualquier quiste presente tiene la misma densidad que el LCR.
TC:
No es el método preferido de diagnóstico, ya que los tumores pequeños pueden quedar ocultos por los artefactos óseos
Una TC sin contraste muestra un nódulo isointenso.
Con contraste: intenso realce homogéneo de los nódulos
La TC también se utiliza como adyuvante de la angiografía cuando el paciente no puede someterse a una RM.
Angiografía cerebral y espinal:
Se observa un elevado “blush” tumoral vascular con arterias agrandadas y venas dilatadas.
Puede detectar el suministro de sangre alALAmyloidosistumorTumorInflammation → ayuda alALAmyloidosis cirujano enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la resección adecuada del tumorTumorInflammation
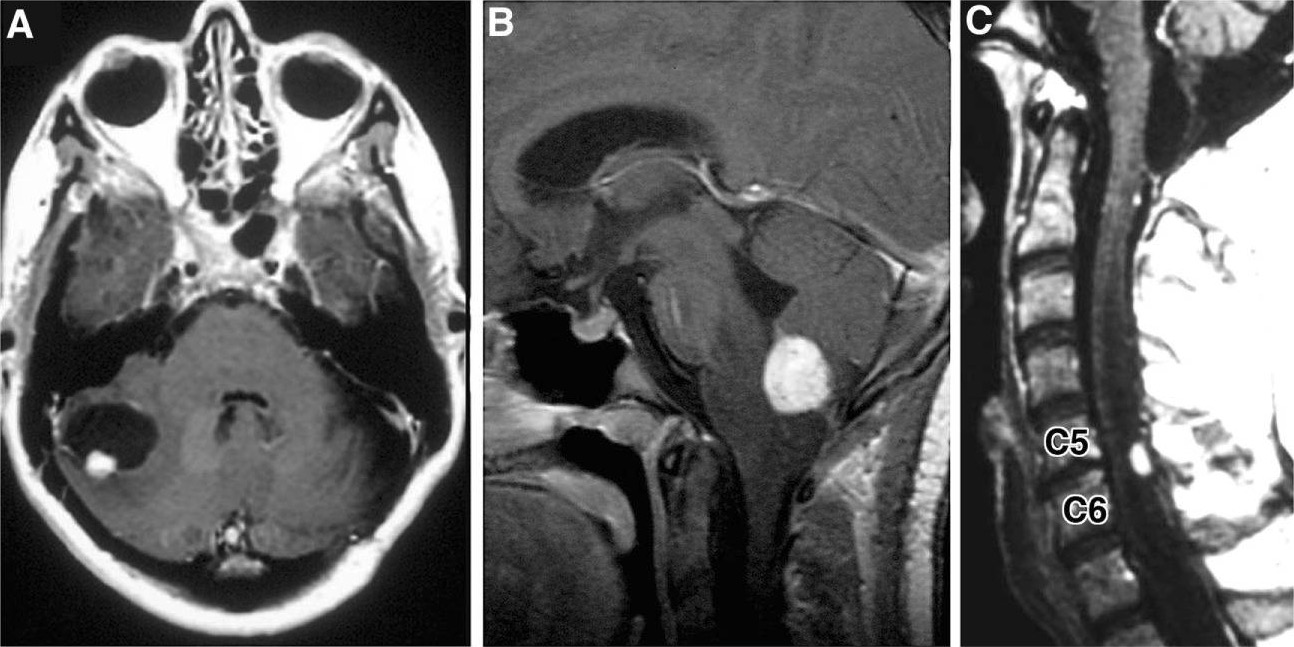
Imágenes radiográficas de hemangioblastomas: A: RM axial potenciada en T1 con contraste que muestra hemangioblastoma cerebeloso con nódulo mural que realza con contraste y quiste peritumoral B: RM sagital potenciada en T1 con contraste que revela hemangioblastoma medular con contraste y edema vasogénico circundante C: RM sagital, realzada con contraste, potenciada en T1 con hemangioblastoma posterior/dorsal realzado con contraste con siringe asociada
Imagen: “Hemangioblastoma” por Department of Neurological Surgery, Ohio State University Wexner Medical Center , Columbus, OH , USA. Licencia: CC BY 4.0
Examen oftalmológico
LosLOSNeisseriahemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease tienen una alta asociación con von Hippel-Lindau, que se asocia con hemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease retinianos.
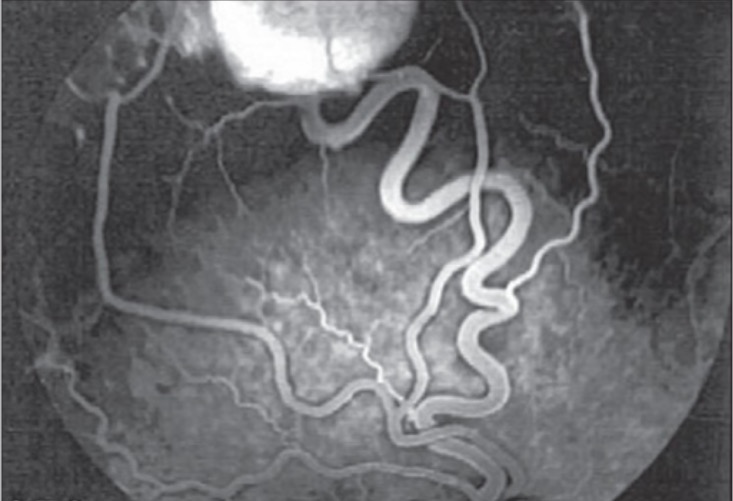
Se puede realizar una angiografía retiniana para descartar la presencia de un hemangioblastomaHemangioblastomaHemangioblastomas are vascular neoplasms of the CNS. Hemangioblastomas are rare and are often associated with von Hippel-Lindau disease (VHL). The most common presentation is a headache and, depending on the size and location of the tumor, patients may present with sensory deficits and motor weakness.Hemangioblastoma retiniano.
Angiografía con fluoresceína del ojo derecho que muestra un hemangioblastoma retiniano superior temporal con la arteria alimentadora y la vena de drenaje
Imagen: “Fluorescein angiogram of right eye” por Vitreoretinal Division, Department of Ophthalmology, Al-Hussein Hospital, King Hussein Medical Center, Amman, Jordan. Licencia: CC BY 2.0
Histopatología
Estándar de oro para el diagnóstico
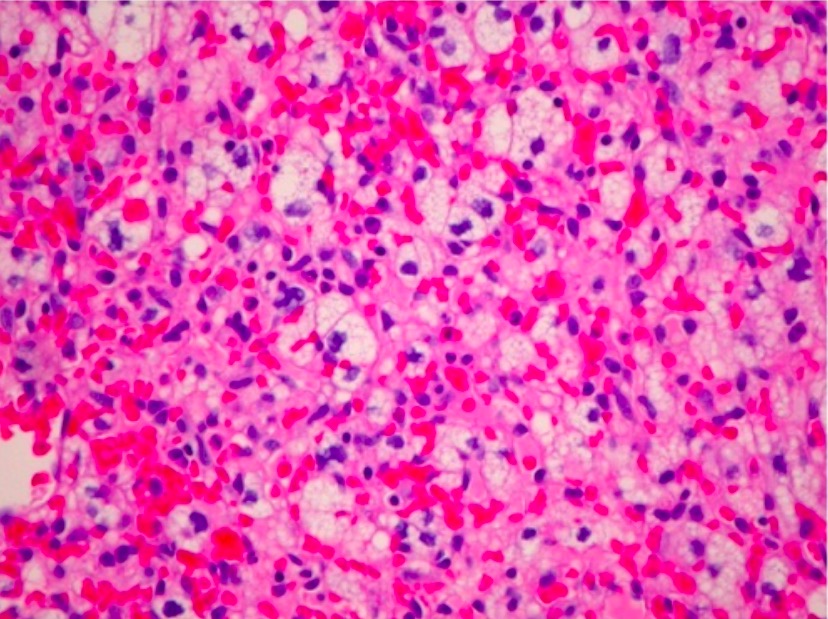
Examen macroscópico: nódulos rojos muy vascularizados bien delimitados
Examinación microscópica:
Amplias redes vasculares con capilares estructuralmente normales
Células estromales neoplásicas:
Núcleos hipercromáticos, pleomórficos
Tasa mitótica baja
Sin atipia
Fibras de Rosenthal
Pueden aparecer 2 componentes celulares distintos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el mismo tumorTumorInflammation:
Tipo 1: pequeñas células endoteliales perivasculares con núcleos hipercromáticos y citoplasma escaso
Tipo 2: citoplasma vacuolado y rico enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum lípidos
Microfotografía H&E de un hemangioblastoma del nervio óptico (ubicación inusual) que revela un tumor marcadamente vascular con células estromales lipidizadas (×200)
Imagen: “Optic nerve hemangioblastoma” por Department of Surgery, Division of Neurosurgery , University of Alabama at Birmingham, Birmingham, AL 35294, USA. Licencia: CC BY 3.0
Tratamiento y Pronóstico
La resección quirúrgica es el enfoque principal para tratar losLOSNeisseriahemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease. Las terapias adyuvantes comúnmente se requieren enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseriahemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease asociados con von Hippel-Lindau, y las opciones incluyen radioterapia, embolización endovascular y terapia antiangiogénica.
Cirugía
La cirugía es el enfoque definitivo primario para tratar losLOSNeisseriahemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease, ya que son tumores benignos no invasivos.
LosLOSNeisseria tumores suelen estar bien delimitados de las estructuras circundantes, pero son muy vascularizados y se localizan enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum áreas neurológicamente sensibles.
La angiografía preoperatoria es útil para identificar las arterias de alimentación.
Necesidad de terapia adyuvante:
Generalmente, no se requiere enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum tumores únicos esporádicos
Típicamente requerida enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum tumores múltiples y/o asociados a von Hippel-Lindau
Otras modalidades terapéuticas
Radioterapia:
La radioterapia postoperatoria reduce la recurrencia de losLOSNeisseriahemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease.
Indicaciones:
Lesiones inaccesibles quirúrgicamente
Múltiples lesiones
Embolización endovascular:
Objetivo: disminuir la vascularización del tumorTumorInflammation y reducir las complicaciones intraoperatorias de la hemorragia
LosLOSNeisseria tumores grandes se pueden reducir considerablemente para facilitar la resección.
Mecanismo de acción: un anticuerpo monoclonal que se une e inhibe losLOSNeisseria efectos del factor de crecimiento endotelial vascular → ↓ angiogénesis
Usos: pacientes con Von Hippel-Lindau con tumores que no responden a la cirugía o a la radiación, o que no son susceptibles de ellas
Inhibidor de HIF-2ɑ (belzutifan):
Para individuos con enfermedad de von Hippel-Lindau conocida
Se utiliza para retrasar la cirugía enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum aquellos con tumores de rápido crecimiento
También es beneficioso para individuos con tumores refractarios o recurrentes
Pronóstico
El pronóstico es bueno enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la mayoría de losLOSNeisseriahemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease tratados:
La resección quirúrgica limpia (con márgenes quirúrgicos negativos), especialmente enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum tumores esporádicos solitarios, asegura un buen pronóstico.
La detección e intervención temprana es favorable.
HemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease asociados con von Hippel-Lindau:
Mayor probabilidad de recurrencia que losLOSNeisseria tumores esporádicos
Peor pronóstico
Mayor asociación con déficits neurológicos
Diagnóstico Diferencial
MeningiomaMeningiomaMeningiomas are slow-growing tumors that arise from the meninges of the brain and spinal cord. The vast majority are benign. These tumors commonly occur in individuals with a history of high doses of skull radiation, head trauma, and neurofibromatosis 2. Meningioma: tumores que surgen de las meningesMeningesThe brain and the spinal cord are enveloped by 3 overlapping layers of connective tissue called the meninges. The layers are, from the most external layer to the most internal layer, the dura mater, arachnoid mater, and pia mater. Between these layers are 3 potential spaces called the epidural, subdural, and subarachnoid spaces. Meninges: Anatomy del cerebro y la médula espinal. LosLOSNeisseria meningiomas suelen ser asintomáticos, pero pueden presentarse con cefalea, convulsiones y alteraciones visuales. El diagnóstico se realiza mediante RM y biopsia. LosLOSNeisseria casos asintomáticos generalmente se mantienen bajo observación, mientras que losLOSNeisseria pacientes sintomáticos se tratan quirúrgicamente o con radiación. A diferencia de losLOSNeisseriahemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease, losLOSNeisseria meningiomas siempre están cerca de las meningesMeningesThe brain and the spinal cord are enveloped by 3 overlapping layers of connective tissue called the meninges. The layers are, from the most external layer to the most internal layer, the dura mater, arachnoid mater, and pia mater. Between these layers are 3 potential spaces called the epidural, subdural, and subarachnoid spaces. Meninges: Anatomy y, a menudo, tienen datos de inserción de la duramadre (e.g., signo de la cola dural).
Glioblastoma multiformeGlioblastoma multiformeGlioblastoma multiforme is a high-grade astrocytoma, an aggressive brain tumor arising from astrocytes, with an unknown cause and a poorly understood link to risk factors. There are two main types: primary, a more aggressive form seen more commonly in older patients, and secondary, developing from lower-grade astrocytomas and seen more commonly in younger patients.Glioblastoma Multiforme: astrocitoma de grado IV según la World Health Organization (WHO) rápidamente progresivo que surge de losLOSNeisseria astrocitos (células gliales enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el cerebro) y se presenta clínicamente como cefalea, náuseas, somnolencia, visión borrosa, cambios de personalidad y convulsiones. La imagenología, la presentación clínica y la biopsia son losLOSNeisseria pilares del diagnóstico. El tratamiento incluye radioterapia, quimioterapia y escisión quirúrgica. El pronóstico es malo, incluso con tratamiento. A diferencia del hemangioblastomaHemangioblastomaHemangioblastomas are vascular neoplasms of the CNS. Hemangioblastomas are rare and are often associated with von Hippel-Lindau disease (VHL). The most common presentation is a headache and, depending on the size and location of the tumor, patients may present with sensory deficits and motor weakness.Hemangioblastoma, el glioblastoma multiformeGlioblastoma multiformeGlioblastoma multiforme is a high-grade astrocytoma, an aggressive brain tumor arising from astrocytes, with an unknown cause and a poorly understood link to risk factors. There are two main types: primary, a more aggressive form seen more commonly in older patients, and secondary, developing from lower-grade astrocytomas and seen more commonly in younger patients.Glioblastoma Multiforme no se asocia con Von Hippel-Lindau.
OligodendrogliomaOligodendrogliomaOligodendrogliomas are malignant CNS tumors arising from neural glial cell precursors. Oligodendrogliomas often arise in the frontal lobes of the brain and have a generally favorable prognosis when compared to other gliomas. Oligodendrogliomas are the 3rd most common CNS tumor. The most frequent presenting symptom is a seizure.Oligodendroglioma:tumorTumorInflammation del SNC que surge de losLOSNeisseria oligodendrocitos. El oligodendrogliomaOligodendrogliomaOligodendrogliomas are malignant CNS tumors arising from neural glial cell precursors. Oligodendrogliomas often arise in the frontal lobes of the brain and have a generally favorable prognosis when compared to other gliomas. Oligodendrogliomas are the 3rd most common CNS tumor. The most frequent presenting symptom is a seizure.Oligodendroglioma puede presentarse con déficits neurológicos focales, convulsiones y cambios de personalidad según la ubicación exacta. El diagnóstico se realiza mediante imagenología de RM y biopsia. LosLOSNeisseria oligodendrogliomas suelen desarrollarse enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria hemisferios cerebrales, normalmente enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el lóbulo frontalFrontalThe bone that forms the frontal aspect of the skull. Its flat part forms the forehead, articulating inferiorly with the nasal bone and the cheek bone on each side of the face.Skull: Anatomy, y rara vez se encuentran enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el área infratentorial o enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la médula espinal. El tratamiento implica la resección quirúrgica posiblemente acompañada de radiación y/o quimioterapia.
Aneurisma cerebral: debilidad enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la pared de un vaso sanguíneo que irriga el cerebro que sobresale y corre el riesgo de romperse. Un aneurisma no roto suele ser asintomático y la ruptura de un aneurisma provoca celafea intensa y repentina. LosLOSNeisseria aneurismas cerebrales se diagnostican con TC, angiografía o ultrasonido. El tratamiento depende de la ubicación y generalmente se trata con un injerto de stent endovascular.
Dengler, V. L., Galbraith, M., Espinosa, J. M. (2014). Transcriptional regulation by hypoxia inducible factors. Critical reviews in biochemistry and molecular biology, 49(1):1–15. Retrieved June 1, 2021, from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4342852/
¡Crea tu cuenta gratis o inicia una sesión para seguir leyendo!
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Medical Premium le brinda acceso completo a todo el contenido y las funciones
Obtenga Premium para ver todos los vídeos
Verifica tu correo electrónico para obtener una prueba gratuita.
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Premium le ofrece acceso completo a todos los contenidos y funciones, incluido el banco de preguntas de Lecturio con preguntas actualizadas de tipo tablero.