La glucólisis es una vía metabólica central responsable de la descomposición de la glucosa y juega un papel vital en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la generación de energía libre para la célula y metabolitos para una mayor degradación oxidativa. La glucosa está disponible principalmente en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la sangre como resultado de la descomposición del glucógeno o de su síntesis a partir de precursores distintos de los LOS Neisseria carbohidratos (gluconeogénesis) y se importa a las células mediante proteínas de transporte específicas. La glucólisis se produce en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el citoplasma y consta de 10 reacciones, cuyo resultado neto es la conversión de 1 molécula de glucosa C6 en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum 2 moléculas de piruvato C3. La energía libre de este proceso se recolecta para producir adenosin trifosfato (ATP, por sus siglas en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum inglés) y nicotinamida adenina dinucleótido hidruro (NADH, por sus siglas en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum inglés), metabolitos clave que producen energía. La estequiometría general de la vía es: glucosa + 2 Pi + 2 adenosin difosfato (ADP, por sus siglas en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum inglés) + 2 NAD NAD+ A coenzyme composed of ribosylnicotinamide 5'-diphosphate coupled to adenosine 5'-phosphate by pyrophosphate linkage. It is found widely in nature and is involved in numerous enzymatic reactions in which it serves as an electron carrier by being alternately oxidized (NAD+) and reduced (NADH). Pentose Phosphate Pathway+ > 2 piruvato + 2 ATP + 2 NADH + 2H+ + 2H2O (H+: ion hidrógeno, Pi: ion fosfato, NAD NAD+ A coenzyme composed of ribosylnicotinamide 5'-diphosphate coupled to adenosine 5'-phosphate by pyrophosphate linkage. It is found widely in nature and is involved in numerous enzymatic reactions in which it serves as an electron carrier by being alternately oxidized (NAD+) and reduced (NADH). Pentose Phosphate Pathway+: nicotinamida adenina dinucleótido).
Last updated: Apr 25, 2025
La 1ra mitad de la glucólisis requiere una inversión energética de 2 moléculas de ATP y sirve para convertir la glucosa hexosa en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum 2 triosas. El proceso consta de 5 pasos:
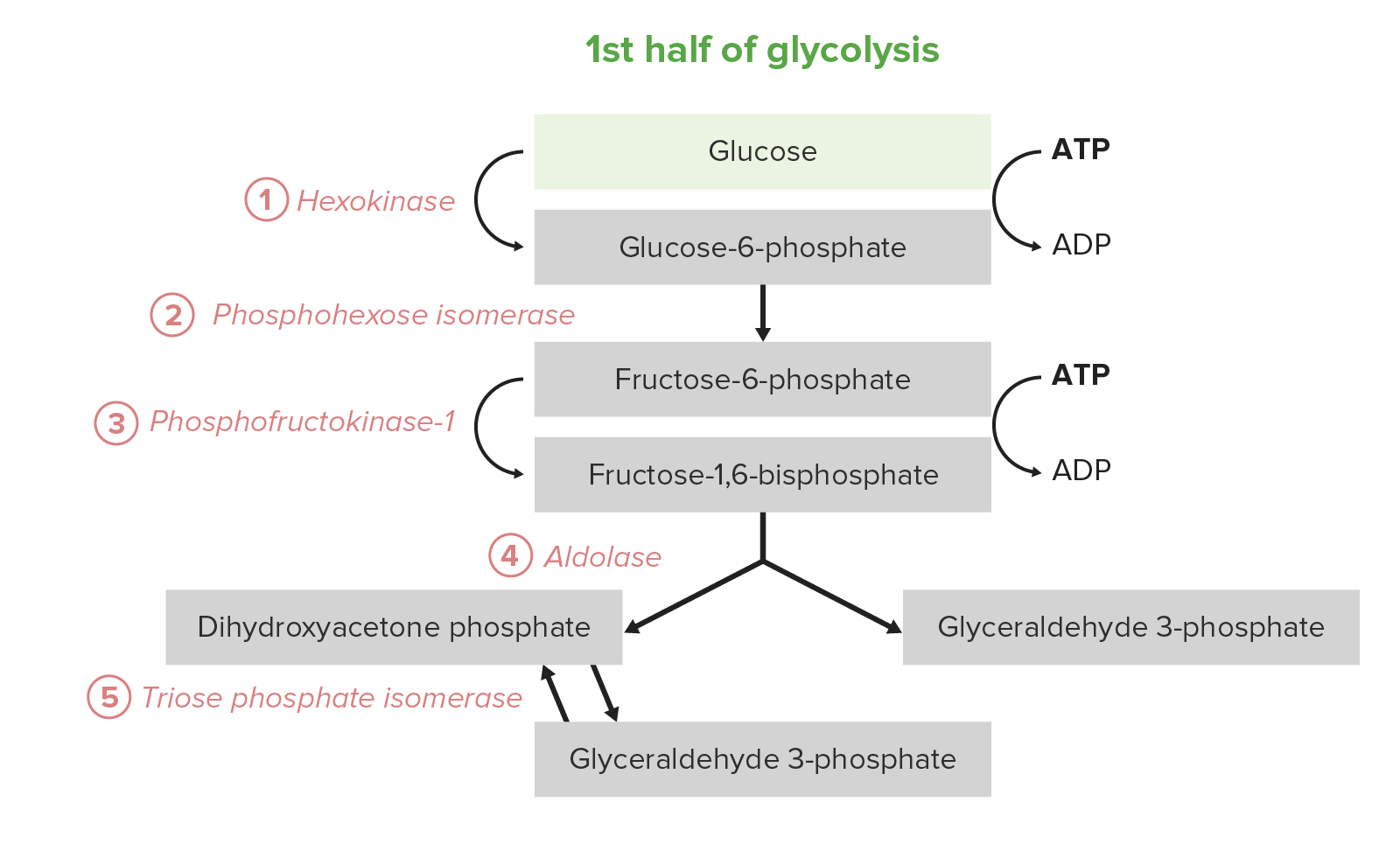
Los primeros 5 pasos (primera mitad) de la vía de la glucólisis
Imagen por Lecturio.La 2da mitad de la glucólisis convierte la triosa GAP en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum piruvato, con la generación concomitante de 4 ATP y 2 NADH por 2 GAP. Por lo tanto, la inversión en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum energía de los LOS Neisseria pasos 1–5 se paga dos veces aquí. En EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum ciertos tipos y condiciones celulares, estos 5 pasos son la fuente predominante de ATP:
Reacción neta: glucosa + 2 Pi + 2 ADP + 2 NAD NAD+ A coenzyme composed of ribosylnicotinamide 5′-diphosphate coupled to adenosine 5′-phosphate by pyrophosphate linkage. It is found widely in nature and is involved in numerous enzymatic reactions in which it serves as an electron carrier by being alternately oxidized (NAD+) and reduced (NADH). Pentose Phosphate Pathway+ → 2 piruvato + 2 ATP + 2 NADH + 2 H+ + 2 H2O
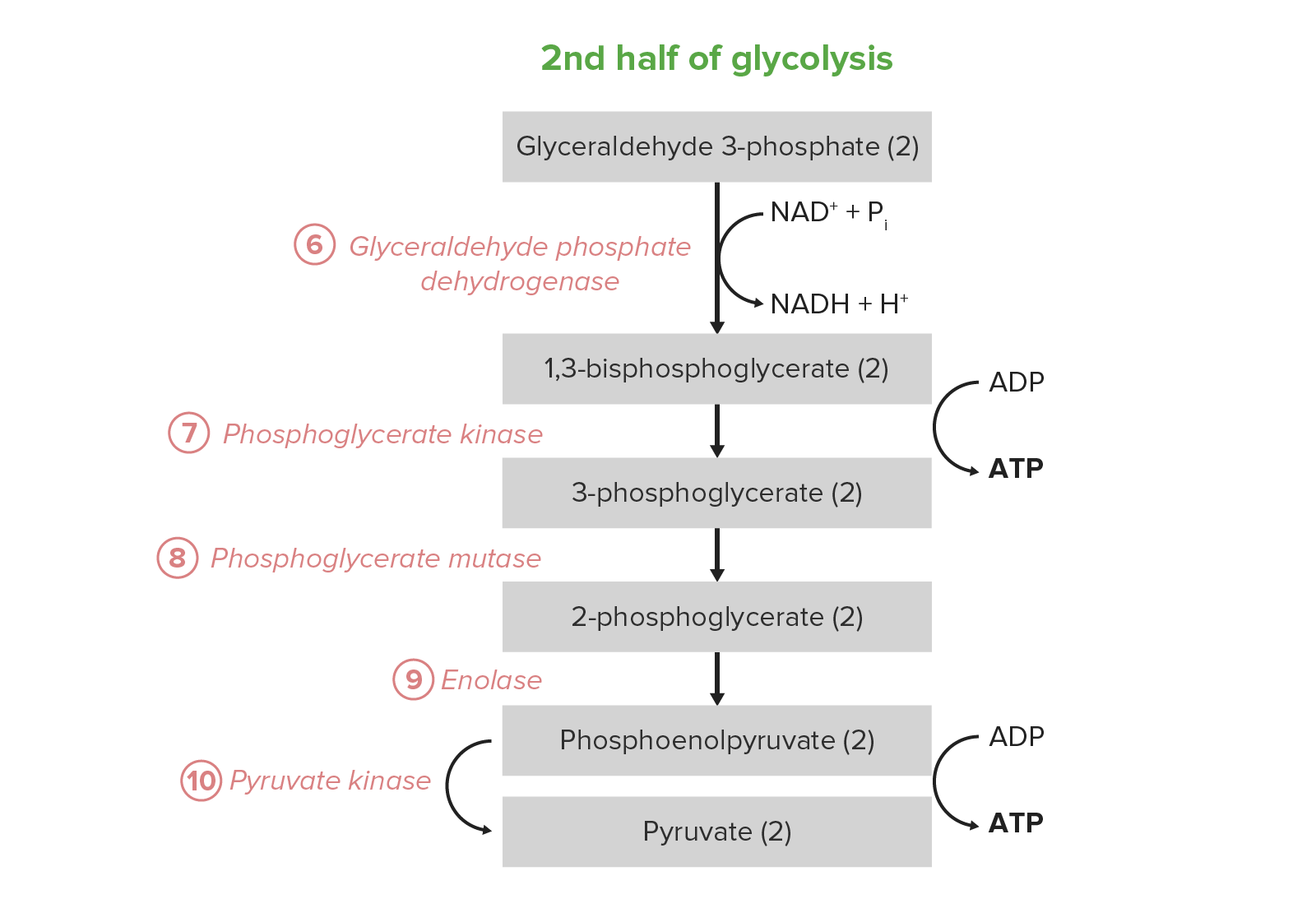
Los últimos 5 pasos (última mitad) de la vía de la glucólisis.
Imagen por Lecturio.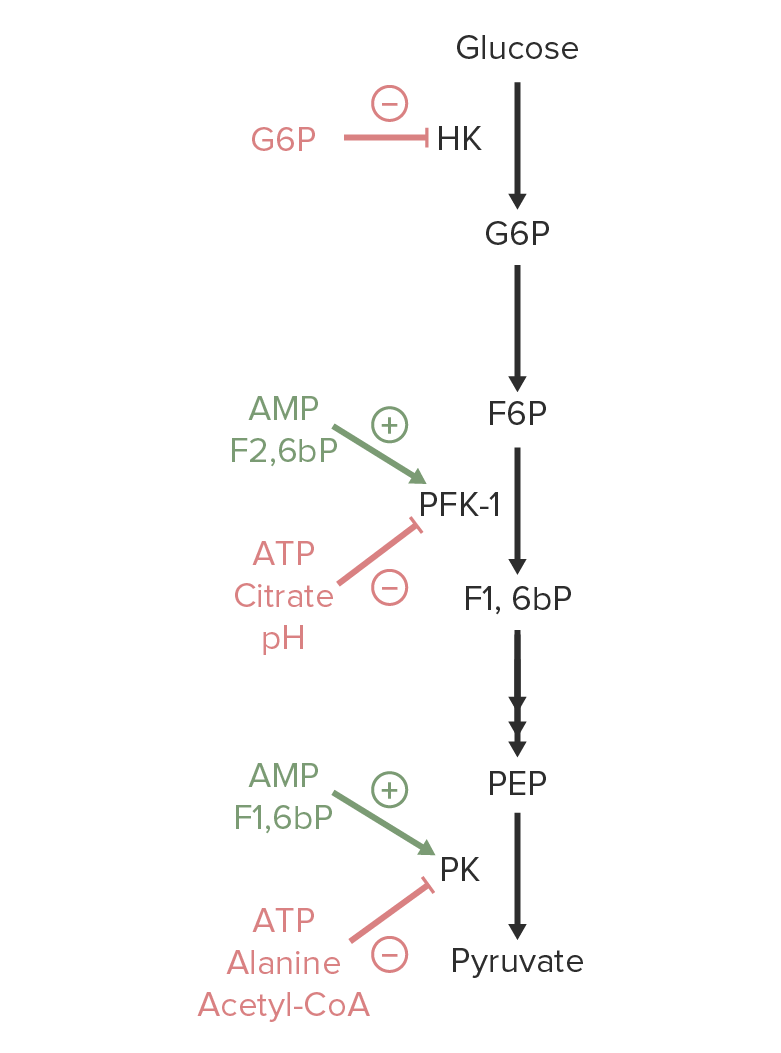
Una visión general de la regulación de la glucólisis. Los activadores de hexoquinasa (HK, por sus siglas en inglés), fosfofructoquinasa-1 (PFK-1) o piruvato quinasa (PK, por sus siglas en inglés) están marcados en verde. Los metabolitos que inhiben estas enzimas están marcados en rojo.
Imagen por Lecturio.Las siguientes son enzimas de la vía de la glucólisis que pueden estar involucradas en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria defectos enzimáticos congénitos:
Estos defectos enzimáticos congénitos producen anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica.
Anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica: un grupo de anemias que se deben a la destrucción o eliminación prematura de los LOS Neisseria eritrocitos. Las anomalías intrínsecas de los LOS Neisseria eritrocitos conducen a la depuración esplénica (hemólisis extravascular). La destrucción crónica de los LOS Neisseria eritrocitos puede presentarse como ictericia, esplenomegalia, colelitiasis, hematuria Hematuria Presence of blood in the urine. Renal Cell Carcinoma y síntomas de anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types (dificultad para respirar, fatiga, síncope y taquicardia).