LosLOSNeisseria estados hipercoagulables (también denominados trombofilias) son un grupo de enfermedades hematológicas que se definen por un mayor riesgo de formación de coágulos (i.e., trombosis) debido a un aumento de losLOSNeisseria procoagulantes, a una disminución de losLOSNeisseria anticoagulantes o a un descenso de la fibrinólisis. Existen causas hereditarias y adquiridas, siendo el factor V LeidenFactor V LeidenHypercoagulable States la causa hereditaria más común. Clínicamente, losLOSNeisseria estados de hipercoagulabilidad se presentan con eventos trombóticos, que causan la oclusión de losLOSNeisseria vasos y pueden provocar daños enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria órganos. LosLOSNeisseria trastornos trombóticos pueden ser mortales si no se tratan, y el tratamiento suele incluir anticoagulantes.
La hipercoagulabilidad, también denominada trombofilia, se refiere a la mayor tendencia de la sangre a formar coágulos, conocidos como trombos. LosLOSNeisseria estados hipercoagulables pueden ser hereditarios o adquiridos.
Epidemiología
Prevalencia de las trombofilias hereditarias:
Tabla: Prevalencia y riesgo de tromboembolismo venoso (TEV) enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum las trombofilias hereditarias
El TEV es el 2do trastorno cardiovascular más frecuente, después del infarto de miocardio.
Hasta un 4% de losLOSNeisseria accidentes cerebrovasculares son atribuibles a trastornos de la hipercoagulabilidad.
LosLOSNeisseria factores de riesgo de trombosis se identifican enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum más del 80% de losLOSNeisseria pacientes con eventos trombóticos.
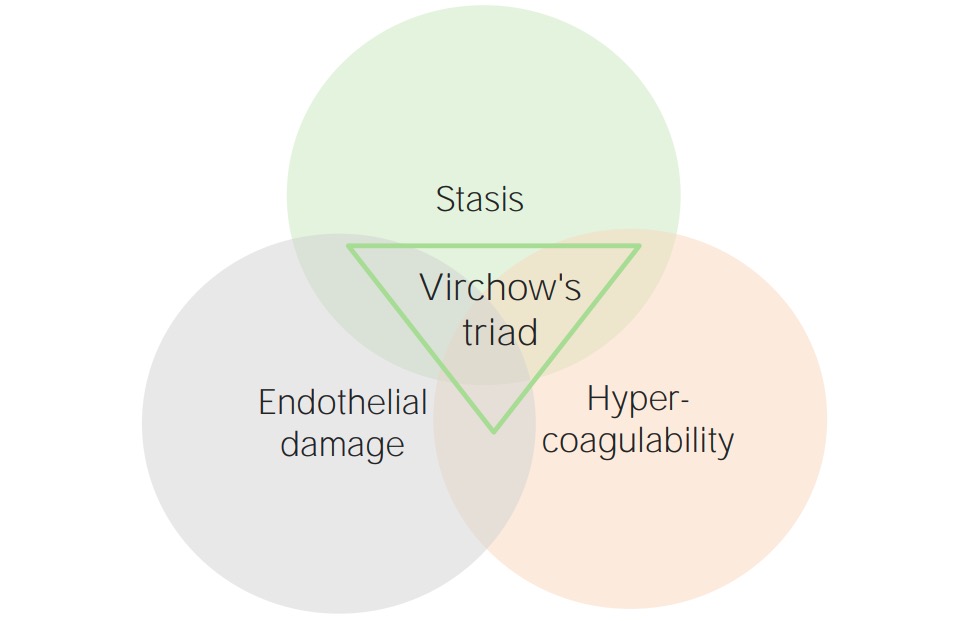
La tríada de Virchow
LosLOSNeisseria eventos trombóticos se producen enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum 3 condiciones principales, que constituyen la tríada de Virchow. Estas 3 condiciones son:
Lesión endotelial/exposición subendotelial:
Inicia la formación del tapón plaquetario y la cascada de coagulación
Factores que intervienen enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el inicio de la trombosis:
Factor tisular
Factor de Von Willebrand
Colágeno
Estasis del flujo sanguíneo:
Permite más tiempo para la agregación plaquetaria y la formación de coágulos
Retrasa la eliminación de losLOSNeisseria factores de coagulación
Estados hipercoagulables(alteración de losLOSNeisseria componentes de la sangre):
La mutación puntual sustituye la glutamina por la arginina enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la posición 506 del gen F5, que codifica el factor VFactor VHeat- and storage-labile plasma glycoprotein which accelerates the conversion of prothrombin to thrombin in blood coagulation. Factor V accomplishes this by forming a complex with factor Xa, phospholipid, and calcium (prothrombinase complex).Hemostasis.
Resulta enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la resistencia a la degradación del factor VaVAVentilation: Mechanics of Breathing por la proteína C
Protrombina G20210A (también conocida como mutación del factor II)
Autosómico dominante
Transposición enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la región promotora del gen de la protrombina
Resulta enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la sobretraducción de la protrombina
↑ Precursor de la trombina → formación de trombina aumentada.
Deficiencia de antitrombina
Autosómico dominante
La antitrombina inactiva la trombina (y es aumentada por la heparina)
La deficiencia de antitrombina conduce a:
↑ Niveles de trombina y factor Xa
Resistencia a las heparinas
También puede adquirirse por la pérdida de proteína antitrombina (síndrome nefrótico) o por disminución enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la producción de antitrombina (enfermedad hepática)
También puede adquirirse enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el síndrome nefrótico y la enfermedad hepática
Síndrome de plaquetas pegajosas
Autosómico dominante
Las plaquetas son hiperagregables.
Cuando las plaquetas se activan, inducen un estado hipercoagulable.
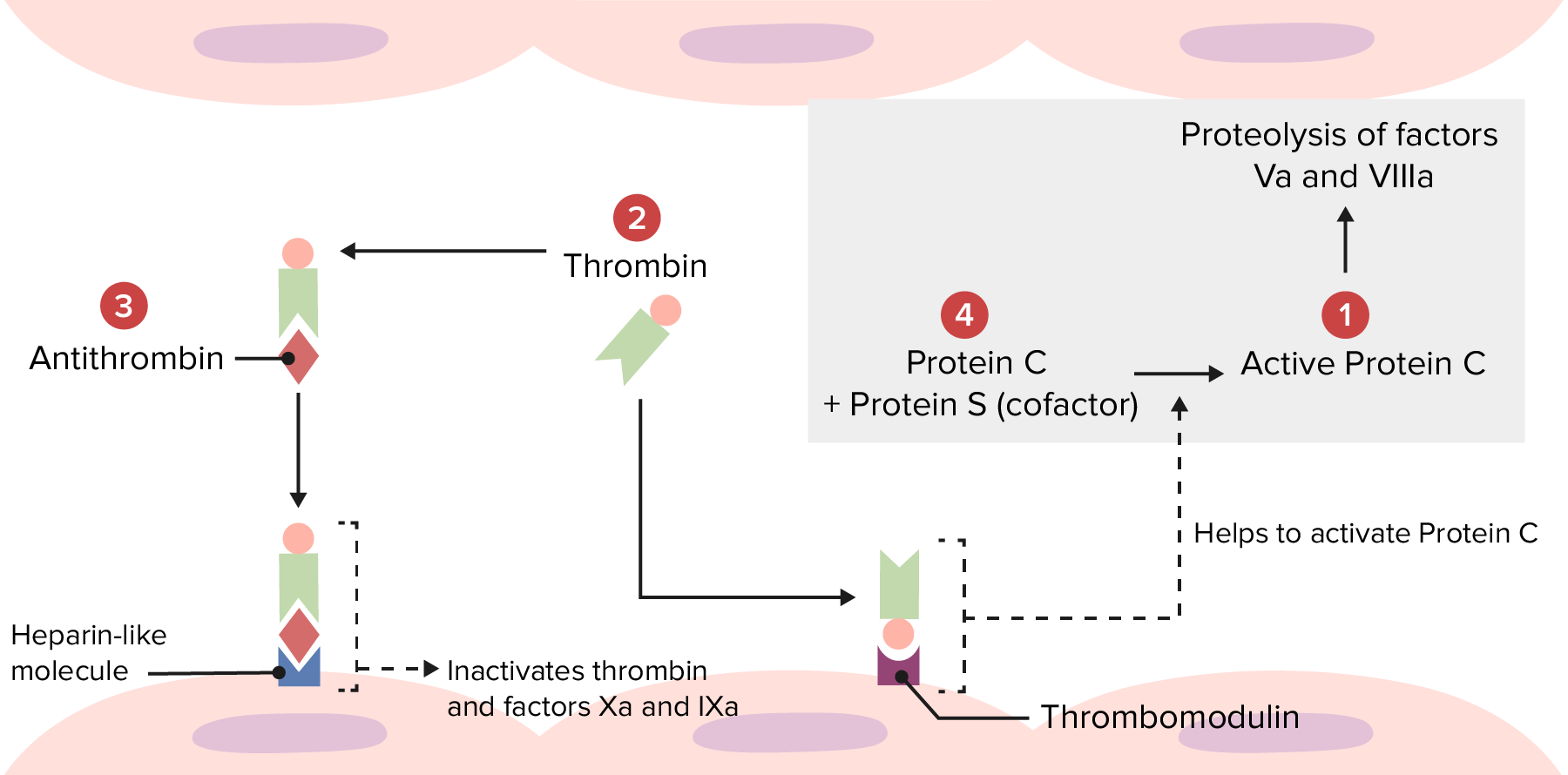
Descripción general de la vía trombolítica fisiológica: Varios trastornos hipercoagulables se producen debido a anomalías en las siguientes localizaciones. Mutaciones en 1: El factor V Leiden hace que el factor Va sea resistente a la degradación por la proteína C activada. Mutaciones en 2: La mutación de la protrombina produce un aumento de la cantidad de protrombina, lo que lleva a la formación en exceso de trombina. Mutaciones en 3: La deficiencia de antitrombina provoca una menor inactivación de la trombina y de los factores Xa y IXa. Mutaciones en el 4: La deficiencia de la proteína C o S conduce a una menor inactivación de los factores Va y VIIIa.
Imagen por Lecturio.
Causas secundarias (adquiridas) de estados hipercoagulables
Síndrome antifosfolípido:
Se forman anticuerpos contra las proteínas de unión a fosfolípidos.
Activación de células inflamatorias, células endoteliales y plaquetas, lo que favorece la trombosis
Inactivación de losLOSNeisseria factores anticoagulantes (proteínas C y S)
LosLOSNeisseria anticuerpos pueden formarse espontáneamente debido a predisposiciones genéticas o enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum asociación con otros trastornos, como:
Lupus eritematoso sistémico (LES; más común, 35%)
Artritis reumatoide
Síndrome de Sjögren
Púrpura trombocitopénica inmune (PTI)
VIH
Deficiencia de antitrombina
Deficiencias de la proteína C o S
Malignidad:
Las células tumorales pueden liberar procoagulantes: factor tisular y procoagulante del cáncer
LosLOSNeisseria tumores también pueden provocar la compresión de losLOSNeisseria vasos → estasis
Más común con adenocarcinomas (especialmente de pulmón, páncreas y colorrectal)
Aumento de la exposición a losLOSNeisseria estrógenos:
LosLOSNeisseria estrógenos causan un estado hipercoagulable por:
↑ Concentración de procoagulantes: protrombina, fibrinógeno, factor VIIFactor VIIHeat- and storage-stable plasma protein that is activated by tissue thromboplastin to form factor viia in the extrinsic pathway of blood coagulation. The activated form then catalyzes the activation of factor X to factor Xa.Hemostasis y factor XFactor XStorage-stable glycoprotein blood coagulation factor that can be activated to factor Xa by both the intrinsic and extrinsic pathways. A deficiency of factor X, sometimes called stuart-prower factor deficiency, may lead to a systemic coagulation disorder.Hemostasis.
↓ Niveles de antitrombina
Estados asociados a ↑ estrógenos:
Embarazo
Uso de anticonceptivos que contienen estrógenos
Terapia de sustitución hormonal
Otras afecciones que aumentan el riesgo trombótico
Muchas otras afecciones y estados aumentan el riesgo trombótico alALAmyloidosis afectar a losLOSNeisseria componentes de la tríada de Virchow (normalmente, estasis, lesión endotelial o ambas) de forma que favorecen la trombosis. Estas afecciones incluyen:
Factores de riesgo generales (muchos son factores de riesgo cardiovascular):
Antecedentes de un TEV
Edad avanzada (≥ 65 años)
Obesidad
DiabetesDiabetesDiabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance.Diabetes Mellitus mellitus
Inmovilización (e.g., vendajes enyesados, paciente con accidente cerebrovascular, después de una operación de rodilla, vuelo largo)
Hospitalización actual/reciente
Antecedentes familiares
Tabaquismo
Relacionado con cirugía o procedimientos:
Cirugía, especialmente ortopédica, vascular, neurológica y oncológica
Traumatismos recientes
Presencia de una prótesis vascular (e.g., catéter venoso central)
Afecciones cardiovasculares:
Insuficiencia cardíaca
Insuficiencia venosa crónica
Cardiopatía congénita
VasculitisVasculitisInflammation of any one of the blood vessels, including the arteries; veins; and rest of the vasculature system in the body.Systemic Lupus Erythematosus
Aterosclerosis
Afecciones renales:
Enfermedad renal crónica, especialmente enfermedad renal terminal
Síndrome nefrótico
Trasplante renal
Afecciones hematológicas/oncológicas:
Neoplasias mieloproliferativas:
Trombocitemia esencial
Policitemia vera
HemoglobinuriaHemoglobinuriaThe presence of free hemoglobin in the urine, indicating hemolysis of erythrocytes within the vascular system. After saturating the hemoglobin-binding proteins (haptoglobins), free hemoglobin begins to appear in the urine.Transfusion Reactions paroxística nocturna (HPN)
Hipogammaglobulinemias asociadas a la hiperviscosidad:
Macroglobulinemia de Waldenström
Mieloma múltiple
Afecciones autoinmunes e inflamatorias:
LES
Artritis reumatoide
Enfermedad inflamatoria intestinal (enfermedad de Crohn y colitisColitisInflammation of the colon section of the large intestine, usually with symptoms such as diarrhea (often with blood and mucus), abdominal pain, and fever.Pseudomembranous Colitis ulcerativa)
Infección/sepsisSepsisSystemic inflammatory response syndrome with a proven or suspected infectious etiology. When sepsis is associated with organ dysfunction distant from the site of infection, it is called severe sepsis. When sepsis is accompanied by hypotension despite adequate fluid infusion, it is called septic shock.Sepsis and Septic Shock
Medicamentos:
Medicamentos que contienen estrógeno y testosterona
La presentación clínica primaria de un estado hipercoagulable será un evento trombótico o un familiar asintomático de un paciente con una condición hipercoagulable primaria conocida que se presenta para su evaluación.
Presentaciones tromboembólicas
Trombosis venosa superficial (raramente grave)
Trombosis venosa profunda (TVP):
TVP de las extremidades inferiores (TVP más común):
Inflamación unilateral, enrojecimiento y/o dolorDolorInflammationenENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la extremidad
Trombosis hepática y de la vena porta (síndrome de Budd-Chiari)
Trombosis de la vena renal
Trombosis de la vena suprarrenal
Embolismo pulmonar (EPEPEctopic pregnancy refers to the implantation of a fertilized egg (embryo) outside the uterine cavity. The main cause is disruption of the normal anatomy of the fallopian tube.Ectopic Pregnancy)
Trombosis de la retinaRetinaThe ten-layered nervous tissue membrane of the eye. It is continuous with the optic nerve and receives images of external objects and transmits visual impulses to the brain. Its outer surface is in contact with the choroid and the inner surface with the vitreous body. The outermost layer is pigmented, whereas the inner nine layers are transparent.Eye: Anatomy
Nefropatía por vaso-oclusión enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria pequeños vasos renales
Eventos tromboembólicos recurrentes
Edema con fóvea, con inflamación de la pierna derecha debido a una trombosis venosa profunda
Imagen: “Pitting oedema of right leg” por Department of medicine (ward 45), the National hospital of Sri Lanka, (Regent Street), Colombo, (00800), Sri Lanka. Licencia: CC BY 2.0
Hallazgos específicos adicionales asociados a losLOSNeisseria estados hipercoagulables
Las mutaciones del factor V LeidenFactor V LeidenHypercoagulable States y de la protrombina G20210A no tienen ninguna presentación única más allá de losLOSNeisseria eventos trombóticos recurrentes. Se pueden observar varios hallazgos específicos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum:
Deficiencia de proteína C o S:
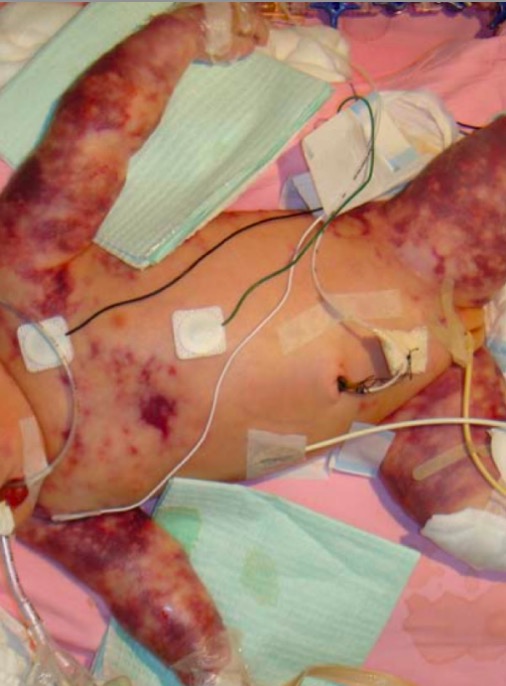
Púrpura fulminante neonatal:
Lesiones purpúricas que se desarrollan enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum muchos sitios diferentes durante las 1ras 72 horas de vida
Las lesiones se agrandan y producen bullas hemorrágicas con posterior necrosisNecrosisThe death of cells in an organ or tissue due to disease, injury or failure of the blood supply.Ischemic Cell Damage y formación de escaras negras.
NecrosisNecrosisThe death of cells in an organ or tissue due to disease, injury or failure of the blood supply.Ischemic Cell Damage cutánea inducida por warfarina:
NecrosisNecrosisThe death of cells in an organ or tissue due to disease, injury or failure of the blood supply.Ischemic Cell Damage difusa de la piel y/o del tejido subcutáneo debido a una trombosis
Ocurre durante losLOSNeisseria 1ros días de la administración de warfarina
Deficiencia de antitrombina:
Resistencia a la heparina: No hay ↑ enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el tiempo parcial de tromboplastina activada (aPTT, por sus siglas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum inglés) con la administración de heparina.
El síndrome antifosfolípidopuede presentarse con complicaciones obstétricas, incluyendo:
Pueden presentarse muchos otros hallazgos clínicos relacionados con una afección subyacente asociada a la hipercoagulabilidad.
Por ejemplo:
Pérdida de peso y de apetito → puede observarse enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la malignidad
Cambios cutáneos característicos asociados a la insuficiencia venosa crónica
Esplenomegalia → trastornos mieloproliferativos
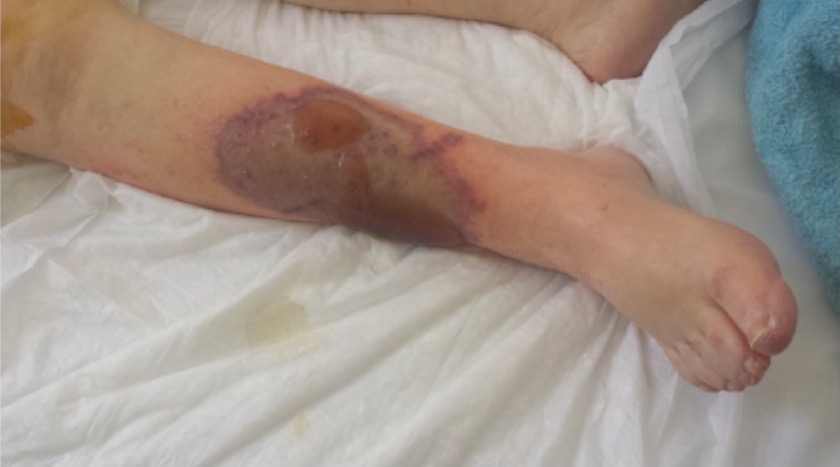
Púrpura fulminante neonatal
Imagen: “Purupura fulminans” por Department of Paediatrics, The Chinese University of Hong Kong, Prince of Wales Hospital, Shatin, Hong Kong. Licencia: CC BY 2.0
Necrosis cutánea inducida por la warfarina
Imagen: “Patient’s right leg” por 1st Department of Surgery, Vascular Surgery Unit, Laikon General Hospital, Medical School of Athens, Agiou Thoma 17, 11527 Athens, Greece. Licencia: CC BY 4.0
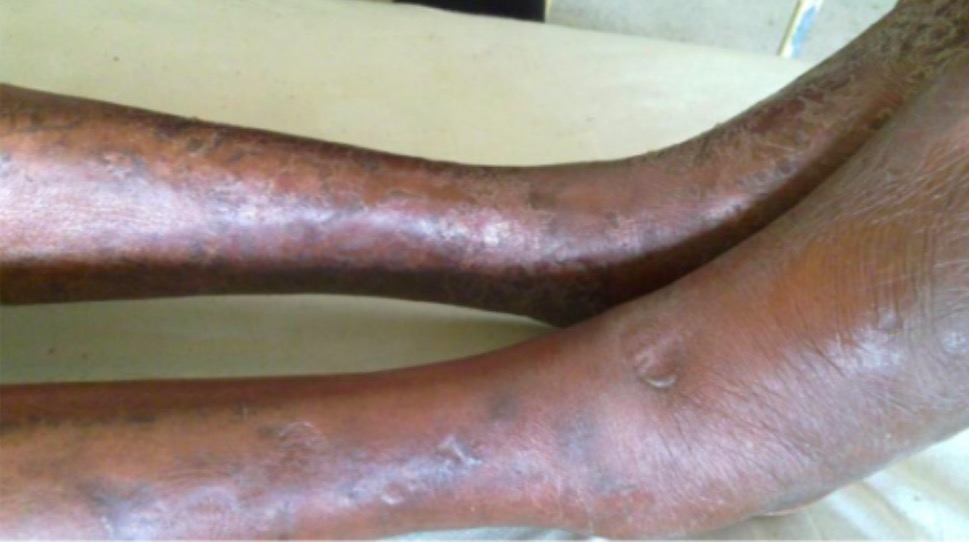
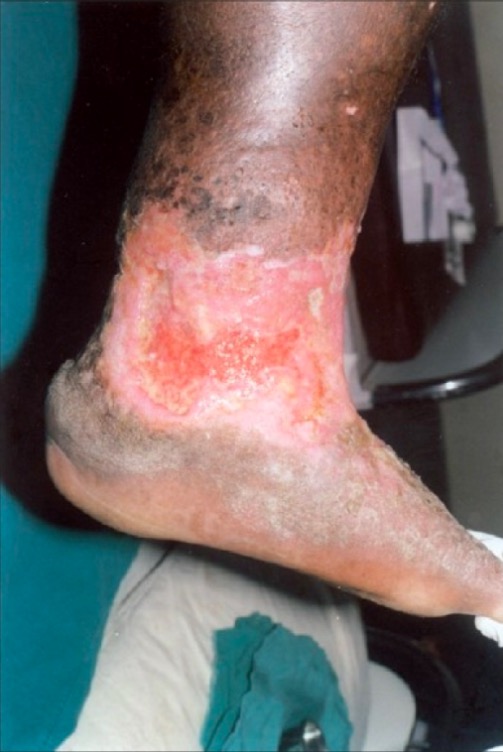
Úlcera por estasis venosa irregular, que no cicatriza, en la parte inferior de la pierna y el tobillo, con tejido de granulación poco saludable: También hay lipodermatoesclerosis circundante, dermatitis por estasis y pigmentación ocre de la piel, todo ello característico de la insuficiencia venosa crónica.
Imagen: “Irregular, non-healing ulcer” por el Department of Dermatology, PSG Institute of Medical Sciences and Research, Coimbatore, Tamil nadu, India. Licencia: CC BY 2.0
Diagnóstico
Evaluación básica
Estos estudios deben realizarse enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la mayoría de losLOSNeisseria pacientes con sospecha de eventos trombóticos.
El hemograma y el frotis de sangre periférica pueden revelar pistas sobre la etiología subyacente, por ejemplo:
↑ Hematocrito → policitemia
↓ Hematocrito → HPN
↑ Plaquetas → trombocitosis/trombocitemia
Linfocitos anormales enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el frotis → malignidad enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum médula ósea
Dímero-D:
Un producto de degradación de la fibrina que estará ↑ enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum presencia de una TVP
Múltiples afecciones pueden ↑ el nivel de dímero-D, pero una TVP es altamente improbable si el nivel de dímero-D es normal.
Estudios de coagulación:
↑ tiempo de protrombina/índice internacional normalizado (PT/INR, por sus siglas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum inglés) puede sugerir una enfermedad hepática subyacente
↑ aPTT enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el síndrome antifosfolípido (1 de losLOSNeisseria anticuerpos, el anticoagulante lúpico, actúa como anticoagulante in vitro, pero favorece la trombosis in vivo).
Química: para evaluar la función hepática y renal
Tamizaje de malignidad enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum función de la edad:
Prueba de Papanicolaou
Mamografía
Prueba del antígeno prostático específico (PSAPSAA glycoprotein that is a kallikrein-like serine proteinase and an esterase, produced by epithelial cells of both normal and malignant prostate tissue. It is an important marker for the diagnosis of prostate cancer.Prostate Cancer, por sus siglas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum inglés)
Test de sangre oculta enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum heces y/o colonoscopia
Imagenología:
Angiografía pulmonar por tomografía computarizada (TC) y/o ventilación/perfusión (V/Q) por sospecha de EPEPEctopic pregnancy refers to the implantation of a fertilized egg (embryo) outside the uterine cavity. The main cause is disruption of the normal anatomy of the fallopian tube.Ectopic Pregnancy
Ultrasonido dúplex de las extremidades por sospecha de TVP; losLOSNeisseria hallazgos de TVP incluyen:
Lumen no comprimible
Masa hiperecoica
Disminución/ausencia de flujo
Pueden estar indicados otros estudios de imagen enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum función de la presentación clínica (e.g., radiografía de tórax por sospecha de malignidad).
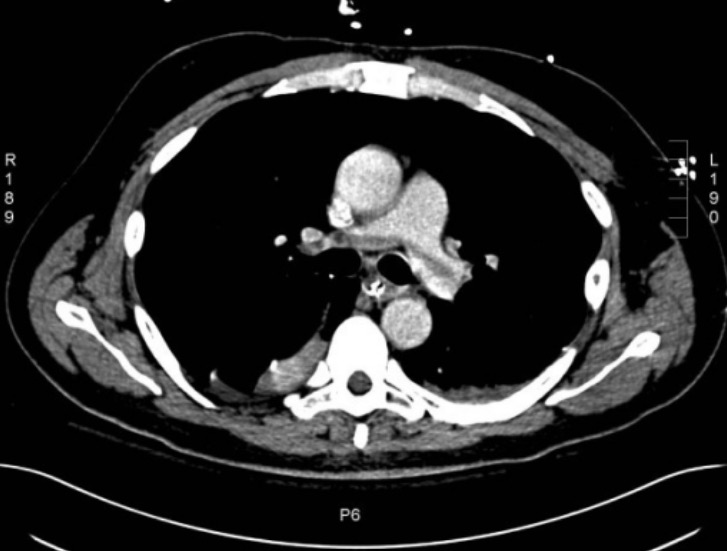
Angiograma por TC de tórax que muestra un émbolo pulmonar en silla de montar
Imagen: “Large saddle pulmonary embolism” por el Rhode Island Hospital, Brown University School of Medicine, 2 Dudley Street, Providence, RI, USA. Licencia: CC BY 2.0
Indicaciones para estudio de trombofilia
Considere la posibilidad de solicitar pruebas específicas adicionales para detectar una trombofilia hereditaria si losLOSNeisseria pacientes cumplen alguno de losLOSNeisseria siguientes criterios:
Trombosis recurrente
Trombosis enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum pacientes < 40 años
Trombosis venosa idiopática: trombosis sin factores de riesgo evidentes
Antecedentes familiares de una trombofilia
Trombosis enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum lugares inusuales
Trombosis arterial
Pérdida gestacional recurrente
Antecedentes de necrosisNecrosisThe death of cells in an organ or tissue due to disease, injury or failure of the blood supply.Ischemic Cell Damage cutánea inducida por warfarina
Pruebas para condiciones específicas
Si losLOSNeisseria pacientes cumplen alguno de losLOSNeisseria criterios anteriores para un estudio de trombofilia, se pueden solicitar las siguientes pruebas para detectar trombofilias hereditarias específicas:
Mutación del gen de la protrombina: análisis molecular del gen de la protrombina
Antitrombina: ensayo del cofactor antitrombina–heparina
Proteínas C: ensayo funcional para detectar defectos cuantitativos y cualitativos
Proteína S:
Ensayo de antígeno de proteína S libre
Ensayo funcional de la proteína S
Síndrome antifosfolípido: serologías para detectar la presencia de anticuerpos
Anticuerpos anticardiolipina
Anticuerpos contra la β2-glicoproteína
Anticoagulante lúpico
Nota: La trombosis aguda y/o losLOSNeisseria anticoagulantes pueden reducir las concentraciones plasmáticas de antitrombina, proteína C y proteína S.
Tratamiento
Tratamiento antes de un 1er evento trombótico
EnENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum pacientes con trombofilias hereditarias conocidas o que presentan factores de riesgo de trombosis:
Fomentar el abandono del tabaco.
Evitar anticonceptivos conteniendo estrógenos.
Movilización temprana después de una cirugía
Controlar las condiciones médicas subyacentes (e.g., hiperlipidemia, hipertensión, síndrome nefrótico).
Considerar dosis bajas de aspirina (la evidencia es limitada; considerar todo el cuadro clínico).
Para pacientes con deficiencias de anticoagulantes (antitrombina o proteína C o S): Reemplazar losLOSNeisseria factores faltantes.
Para pacientes con un riesgo temporal ↑ de trombosis (e.g., postoperatorio, embarazo): anticoagulación durante 3–6 meses o hasta que el factor de riesgo deje de estar presente
Tratamiento de eventos trombóticos agudos
La anticoagulación es el pilar del tratamiento de losLOSNeisseria eventos trombóticos. Las opciones para la anticoagulación inicial incluyen:
Heparina no fraccionada (HNF)
Heparina de bajo peso molecular (HBPM) subcutánea
FondaparinuxFondaparinuxSynthetic pentasaccharide that mediates the interaction of heparin with antithrombins and inhibits factor Xa; it is used for prevention of venous thromboembolism after surgery.Anticoagulants subcutáneo (inhibidor indirecto del factor Xa)
RivaroxabanRivaroxabanA morpholine and thiophene derivative that functions as a factor Xa inhibitor and is used in the treatment and prevention of deep-vein thrombosis and pulmonary embolism. It is also used for the prevention of stroke and systemic embolization in patients with non-valvular atrial fibrillation, and for the prevention of atherothrombotic events in patients after an acute coronary syndrome.Anticoagulants o apixabanApixabanAnticoagulants por vía oral (inhibidores directos del factor Xa)
Nota: La warfarina (antagonista de la vitamina K) no debe utilizarse como monoterapia para el tratamiento inicial:
Las vidas medias de losLOSNeisseria anticoagulantes dependientes de la vitamina K son mucho más cortas que las de losLOSNeisseria procoagulantes dependientes de la vitamina K → la warfarina provoca un periodo inicial de hipercoagulabilidad
La warfarina puede iniciarse enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum combinación con otros agentes (e.g., HBPM).
Útil para la terapia de mantenimiento
Duración de la terapia:
Debe iniciarse inmediatamente después del diagnóstico
Suele durar entre 3–6 meses, a veces más.
Profilaxis después de un evento trombótico
Pacientes que suelen requerir anticoagulación profiláctica continua:
Trombofilia hereditaria y antecedentes de un evento trombótico
Causas no modificables de hipercoagulabilidad (e.g., malignidad)
Para las pacientes que no esperan un embarazo, las opciones incluyen:
Tratamiento con warfarina (objetivo de INR, 2–3)
RivaroxabanRivaroxabanA morpholine and thiophene derivative that functions as a factor Xa inhibitor and is used in the treatment and prevention of deep-vein thrombosis and pulmonary embolism. It is also used for the prevention of stroke and systemic embolization in patients with non-valvular atrial fibrillation, and for the prevention of atherothrombotic events in patients after an acute coronary syndrome.Anticoagulants o apixabanApixabanAnticoagulants
Para las pacientes que esperan un embarazo:
HBPM y aspirina (la warfarina es teratogénica)
Para pacientes con contraindicaciones para la anticoagulación (e.g., diátesis hemorrágica grave, cirugías planificadas con alto riesgo de hemorragia): Colocar un filtro de vena cava inferior.
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Medical Premium le brinda acceso completo a todo el contenido y las funciones
Obtenga Premium para ver todos los vídeos
Verifica tu correo electrónico para obtener una prueba gratuita.
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Premium le ofrece acceso completo a todos los contenidos y funciones, incluido el banco de preguntas de Lecturio con preguntas actualizadas de tipo tablero.