La deficiencia de piruvato quinasa (PK) es un trastorno enzimático autosómico recesivo de losLOSNeisseria eritrocitos que provoca anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica. La deficiencia de PK es principalmente un trastorno hereditario, con un defecto enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el gen PKLR, pero puede adquirirse de forma secundaria a afecciones subyacentes, como la leucemia. Las características clínicas típicas de la deficiencia de PK son anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types de leve a moderada, ictericia, retraso del crecimiento, retraso enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el desarrollo y protuberancia frontalFrontalThe bone that forms the frontal aspect of the skull. Its flat part forms the forehead, articulating inferiorly with the nasal bone and the cheek bone on each side of the face.Skull: Anatomy. Sin embargo, son posibles presentaciones más severas, especialmente enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum pacientes jóvenes. El diagnóstico implica descartar otras causas más frecuentes de anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica. Las pruebas bioquímicas y/o genéticas pueden confirmar el diagnóstico. El manejo puede incluir tratamiento de soporte, mitapivat (un activador de PK recientemente aprobado por la FDA), exanguinotransfusión, fototerapia o esplenectomía. El pronóstico depende de la edad, asociándose una mayor edad enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el momento del diagnóstico con un mejor pronóstico.
La deficiencia de piruvato quinasa (PK) es un trastorno enzimático autosómico recesivo de losLOSNeisseria eritrocitos que provoca hemólisis crónica debido a un defecto enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el gen PKLR.
Epidemiología
Defecto extremadamente raro, pero el más común enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la vía de la glucólisis.
La prevalencia se desconoce; las estimaciones han oscilado entre 1 de cada 20 000 y 1 de cada 115 000.
Distribución mundial
Más común enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum:
Personas de ascendencia noreuropea y china
Poblaciones aisladas con antecedentes de consanguinidad por efecto fundador
Hereditaria (mutaciones genéticas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el gen PKLR):
LosLOSNeisseria individuos heterocigotos suelen ser asintomáticos.
Gen PKLR localizado enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el cromosoma 1q21
Adquirida (secundaria a enfermedades subyacentes):
Leucemia
AnemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types sideroblástica refractaria
Complicación de quimioterapia
Fisiología normal
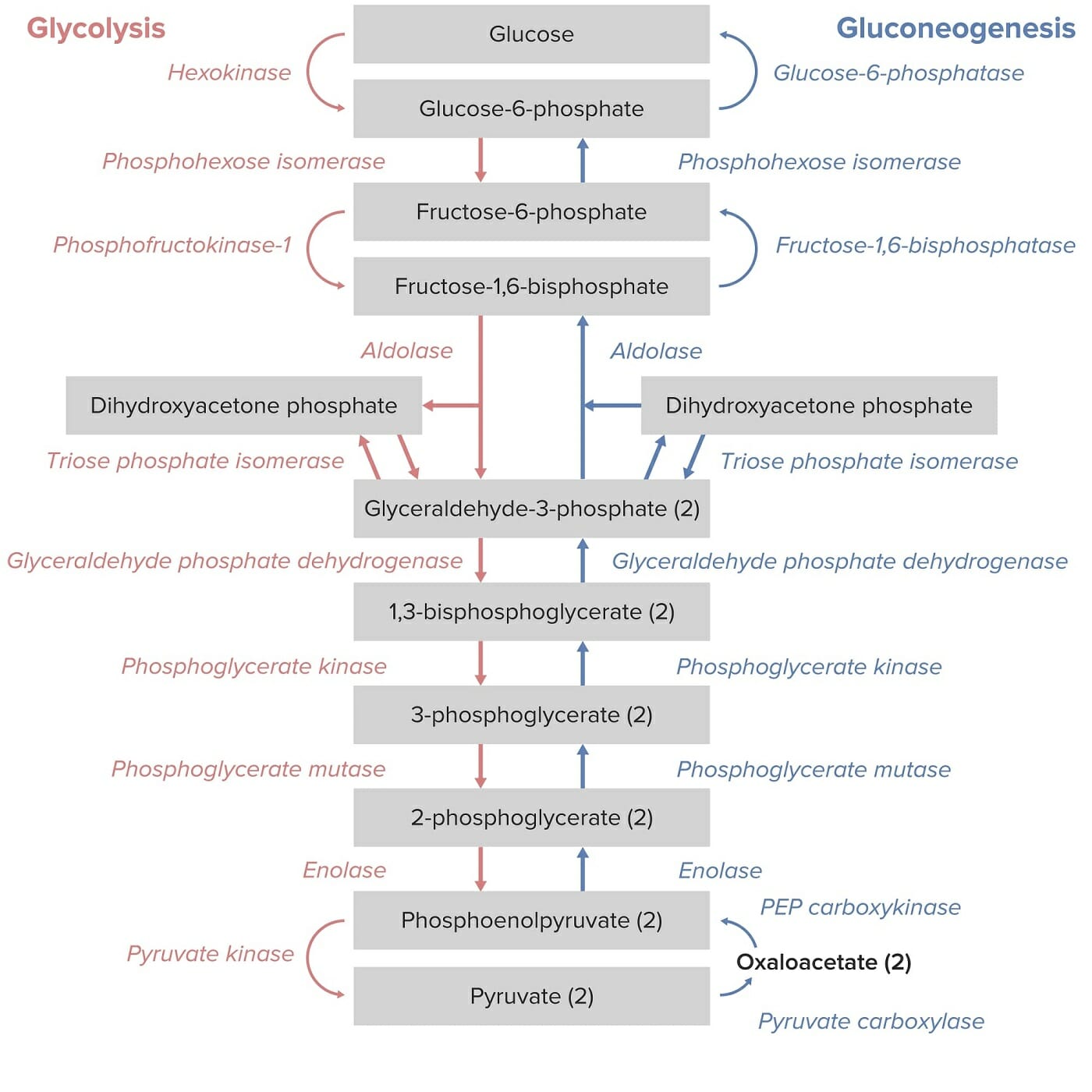
La piruvato quinasa es una enzima necesaria para la conversión de fosfoenolpiruvato (PEPPEPA monocarboxylic acid anion derived from selective deprotonation of the carboxy group of phosphoenolpyruvic acid. It is a metabolic intermediate in glycolysis; gluconeogenesis; and other pathways.Glycolysis, por sus siglas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum inglés) enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum piruvato y ATP enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la vía de la glucólisis, que produce energía.
LosLOSNeisseria eritrocitos dependen de la glucólisis para producir ATP (carecen de mitocondrias).
El ATP es necesario para el mantenimiento de la función celular y la integridad estructural.
Vías de la glucólisis y la gluconeogénesis: Obsérvese que la conversión de fosfoenolpiruvato en piruvato por la piruvato quinasa es el paso final de la vía de la glucólisis.
Imagen por Lecturio.
Fisiopatología
Efecto de la deficiencia de PK:
Acumulación de precursores enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la vía de la glucólisis, especialmente 2,3-difosfoglicerato (2,3-DPG).
↓ Producción de:
Piruvato
ATP
Lactato
Consecuencias de la deficiencia de PK:
Hemólisis:
Acumulación de precursores intermedios y metabolitos glucolíticos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria eritrocitos:
↓ Plasticidad de la membrana eritrocitaria y deshidratación celular → destrucción prematura de eritrocitos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el bazo.
La hemólisis suele ser extravascular, pero la hemólisis severa puede ser intravascular.
↑ Suministro de oxígeno:
↑ 2,3-DPG → desplazamiento hacia la derecha de la curva de disociación del oxígeno (a los eritrocitos les cuesta más “retener” el O2) → resulta enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum ↑ aporte de oxígeno a losLOSNeisseria tejidos periféricos por volumen de hemoglobina.
Permite una mayor tolerancia fisiológica a la anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types con deficiencia de PK
Sobrecarga de hierro:
Eritropoyesis inefectiva → ↑ producción de precursores eritrocitarios precoces.
↓ Hepcidina, que conduce a:
↑ Absorción de hierro
↓ Retención de hierro enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum macrófagos
Presentación Clínica y Complicaciones
Presentación clínica
La edad y la severidad enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el momento de la presentación dependen de la extensión de la hemólisis y la anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types. Además, la edad de presentación puede variar.
Signos y síntomas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum recién nacidos:
Ictericia neonatal/kernícterus
Retraso enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el crecimiento, escaso aumento de peso
Retraso del crecimiento, letargo
Protuberancia frontalFrontalThe bone that forms the frontal aspect of the skull. Its flat part forms the forehead, articulating inferiorly with the nasal bone and the cheek bone on each side of the face.Skull: Anatomy
Hematopoyesis extramedular cutánea
Hidropesía fetal (debida a una anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types severa in utero)
Signos y síntomas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum niños más grandes y adultos:
Hallazgos compatibles con anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica crónica.
Palidez
Ictericia
Esplenomegalia de leve a grave
Cálculos biliares pigmentarios (bilirrubina)
Úlceras crónicas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum las piernas
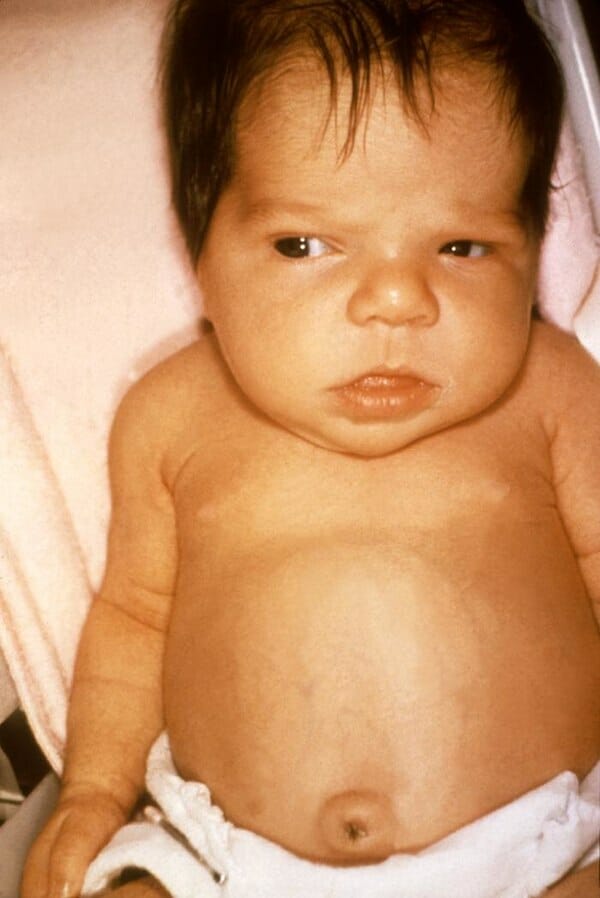
Niña de 6 semanas con síntomas de ictericia por hipotiroidismo.
Imagen: “Jaundice in the newborn” por el Dr. Hudson (CDC). Licencia: Dominio Público
Complicaciones
Insuficiencia cardíaca
Hepatopatía
Disfunción endocrina (por ejemplo, diabetesDiabetesDiabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance.Diabetes Mellitus mellitus, hipogonadismo)
Artropatías
SepsisSepsisSystemic inflammatory response syndrome with a proven or suspected infectious etiology. When sepsis is associated with organ dysfunction distant from the site of infection, it is called severe sepsis. When sepsis is accompanied by hypotension despite adequate fluid infusion, it is called septic shock.Sepsis and Septic Shock postesplenectomía (mayor incidencia enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum niños)
Enfermedad tromboembólica postesplenectomía (mayor incidencia enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum adultos)
Hipertensión pulmonar
Mayor riesgo de anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types aplásica con ciertas infecciones virales (por ejemplo, parvovirus B19Parvovirus B19Primate erythroparvovirus 1 (generally referred to as parvovirus B19, B19 virus, or sometimes erythrovirus B19) ranks among the smallest DNA viruses. Parvovirus B19 is of the family Parvoviridae and genus Erythrovirus. In immunocompetent humans, parvovirus B19 classically results in erythema infectiosum (5th disease) or “slapped cheek syndrome.”Parvovirus B19).
Accidente cerebrovascular isquémico
Riesgos asociados a transfusiones sanguíneas repetidas
Diagnóstico
La deficiencia de piruvato quinasa es rara; es importante descartar otras causas de anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica.
Antecedentes y examen físico
Características clínicas de la anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica
Antecedentes familiares de:
Síntomas similares
Causa desconocida de la anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types
Deficiencia de piruvato quinasa
Evaluación básica de laboratorio
Hemograma completo con diferencial:
↓ Hemoglobina y hematocrito
Volumen corpuscular medio (VCM) normal o ↑.
↑ Reticulocitos (especialmente después de una esplenectomía)
Plaquetas normales o ↑ (trombocitosis reactiva)
Química:
↑ Bilirrubina indirecta
↑ Lactato deshidrogenasa (LDHLDHOsteosarcoma; puede ser normal)
↓ Haptoglobina
Pruebas de antiglobulina:
Prueba de antiglobulina directa (también conocida como prueba de Coombs) negativa
Prueba de antiglobulina indirecta negativa
Frotis de sangre periférica, centrándose enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la morfología de losLOSNeisseria eritrocitos:
LosLOSNeisseria hallazgos característicos de otras causas de anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica hereditaria están ausentes.
Células espinosas (equinocitos)
Eritropoyesis acelerada:
Policromasia
Anisocitosis
Poiquilocitosis
Eritrocitos nucleados
Pruebas confirmatorias
El diagnóstico se confirma si un paciente con anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica/hemólisis compensada presenta anomalías enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum las pruebas bioquímicas o genéticas.
Pruebas bioquímicas:
Estándar de oro
Evalúa la actividad de la piruvato quinasa enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria eritrocitos
Se espera ↓ actividad piruvato quinasa enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la deficiencia de PK.
Un resultado negativo no elimina la posibilidad de una deficiencia de PK:
LosLOSNeisseria leucocitos y las plaquetas también tienen actividad PK. Si no se eliminan del hemolisado antes de la prueba, losLOSNeisseria resultados pueden estar sesgados.
Si se sigue sospechando una deficiencia de PK, considere la posibilidad de realizar ensayos especializados o pruebas genéticas.
Nota: Las pruebas bioquímicas deben aplazarse 2-3 meses después de una transfusión.
Pruebas genéticas:
Busca mutaciones enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumPKLR
No se realizan como pruebas iniciales; la importancia de algunas mutaciones no está clara.
Indicaciones de las pruebas genéticas:
Un individuo con hemólisis o anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types inexplicable que tiene un hermano con una deficiencia conocida de PK.
Pacientes con anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica con una prueba negativa de antiglobulina directa cuando se han descartado otras causas.
Pruebas prenatales a embarazadas que ya tienen un hijo con:
Hidropesía fetal
AnemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types severa dependiente de transfusión, no resuelta por esplenectomía
Tratamiento y Pronóstico
Tratamiento
El tratamiento depende de la edad enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el momento del diagnóstico y de la severidad.
Periodo intrauterino (enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum caso de hidropesía fetal por anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types severa): transfusión intrauterina.
Periodo neonatal (presencia de hiperbilirrubinemia):
Fototerapia
Exanguinotransfusión
Desde la infancia hasta la adultez (presencia de anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types severa):
Transfusiones de eritrocitos
Farmacoterapia:
Mitapivat: un activador de la piruvato quinasa recientemente aprobado por la FDA para el tratamiento de la anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica sintomática.
Ácido fólico
Esplenectomía: indicada enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum casos severos, dependientes de transfusión, que no responden a la mitapivat
Quelación del hierro (sobre todo enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria casos que requieren tratamiento a largo plazo con transfusiones)
Terapias enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum investigación:
Trasplante de células madre hematopoyéticas (para pacientes con anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types severa que requieren múltiples transfusiones tras una esplenectomía).
Terapia génica
Monitoreo
Se debe monitorear enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria pacientes lo siguiente:
Síntomas y signos clínicos de anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types
Crecimiento y desarrollo
Laboratorios:
Hemograma completo con diferencial
Recuento de reticulocitos
Niveles de ferritina
Pronóstico
Formas leves y moderadas de deficiencia de PK: pronóstico excelente
Formas severas de deficiencia de PK: pronóstico dependiente de la edad
Principalmente sintomático enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la primera infancia
Cuanto mayor es el paciente enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el momento del diagnóstico, mejor es el pronóstico.
Diagnóstico Diferencial
Deficiencia de glucosa-6-fosfato deshidrogenasa (G6PDG6PDPentose Phosphate Pathway): Trastorno hereditario recesivo ligado alALAmyloidosis cromosoma X que causa anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica debido a deficiencias enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la enzima G6PDG6PDPentose Phosphate Pathway, que normalmente ayuda a losLOSNeisseria eritrocitos a generar NADPHNADPHNicotinamide adenine dinucleotide phosphate. A coenzyme composed of ribosylnicotinamide 5′-phosphate (nmn) coupled by pyrophosphate linkage to the 5′-phosphate adenosine 2.Pentose Phosphate Pathway y losLOSNeisseria protege de las lesiones oxidativas. Se presenta con síntomas de anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica, como ictericia, esplenomegalia y palidez. El diagnóstico se realiza mediante la presentación clínica, el análisis de frotis sanguíneo y la medición del nivel de enzima G6PDG6PDPentose Phosphate Pathway. El tratamiento es sintomático e incluye evitar losLOSNeisseria desencadenantes.
AnemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types drepanocítica o de células falciformes: grupo de trastornos genéticos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria que una molécula anómala de hemoglobina transforma losLOSNeisseria eritrocitos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum células falciformes. Esto provoca anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types crónica, episodios vaso-oclusivos, dolorDolorInflammation y daños orgánicos. LosLOSNeisseria pacientes son susceptibles de sufrir infecciones, infartos de diversos órganos y aplasiaAplasiaCranial Nerve Palsies medular. La afectación pulmonar enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el síndrome torácico agudo puede ser rápidamente mortal. Por lo general, las células falciformes pueden verse enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el frotis periférico, pero se necesita una electroforesis de hemoglobina para el diagnóstico. El manejo incluye tratamiento sintomático, hidroxiurea, eliminación de desencadenantes y analgésicos.
Hiperbilirrubinemia del recién nacido: elevación de la bilirrubina sérica total, que se presenta clínicamente con una coloración amarillenta de la piel, la esclerótica y las mucosas. Aunque este trastorno es típicamente ictericia fisiológica, debe distinguirse de la ictericia patológica. El diagnóstico se basa enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria hallazgos clínicos y losLOSNeisseria análisis de sangre. El tratamiento puede incluir fototerapia y exanguinotransfusión.
Esferocitosis hereditaria: trastorno causado por una deficiencia de proteínas citoesqueléticas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la membrana de losLOSNeisseria eritrocitos. Esto provoca la pérdida de estabilidad de la membrana y la deformabilidad del eritrocito, lo que da a la célula una forma esférica (esferocito). Estas células están predispuestas a la degradación esplénica, lo que conduce a la hemólisis. La enfermedad se presenta con anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types de leve a moderada, hiperbilirrubinemia, reticulocitosis, aumento de la CHCM y esferocitos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el frotis de sangre periférica. El tratamiento incluye ácido fólico y esplenectomía (tratamiento definitivo).
Referencias
Bianchi, P., et al. (2018). Addressing the diagnostic gaps in pyruvate kinase deficiency: consensus recommendations on the diagnosis of pyruvate kinase deficiency. American Journal of Hematology, 94, 149‒161. https://doi.org/10.1002/ajh.25325
Grace, R. F., Barcellini, W. (2020). Management of pyruvate kinase deficiency in children and adults. Blood, 136(11), 1241–1249. https://doi.org/10.1182/blood.2019000945
Al-Samkari, H., Galactéros, F., et al. (2022). Mitapivat versus placebo for pyruvate kinase deficiency. New England Journal of Medicine, 386(15), 1432–1442. https://doi.org/10.1056/NEJMoa2116634
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Medical Premium le brinda acceso completo a todo el contenido y las funciones
Obtenga Premium para ver todos los vídeos
Verifica tu correo electrónico para obtener una prueba gratuita.
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Premium le ofrece acceso completo a todos los contenidos y funciones, incluido el banco de preguntas de Lecturio con preguntas actualizadas de tipo tablero.