LosLOSNeisseria craneofaringiomas son tumores epiteliales escamosos intracraneales poco frecuentes con una estructura sólida y/o quística que surgen de losLOSNeisseria remanentes de la bolsa de Rathke a lo largo del tallo hipofisario, enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la región supraselar. LosLOSNeisseria craneofaringiomas son histológicamente benignos, pero tienden a invadir las estructuras circundantes; por lo tanto, deben tratarse como enfermedades malignas de bajo grado. Histológicamente, existen 2 tipos de tumores: el adamantinomatoso, que es más común enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria niños; y el papilar, que tiende a presentarse más enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria adultos. Ambos tipos pueden presentarse con diversos síntomas dependiendo de su localización y patrones de extensión. LosLOSNeisseria síntomas incluyen cefaleas, náuseas, vómitos, alteraciones visuales, disfunción endocrina y problemas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el comportamiento. El diagnóstico se realiza mediante la imagenología e histología. El tratamiento suele incluir la escisión quirúrgica y la radioterapia.
LosLOSNeisseria craneofaringiomas son tumores epiteliales escamosos poco frecuentes con una estructura sólida y/o quística, que surgen de losLOSNeisseria remanentes de la bolsa de Rathke a lo largo del tallo hipofisario, enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum línea desde la nasofaringe hasta el diencéfalo (enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la región supraselar).
LosLOSNeisseria craneofaringiomas tienen una histología benigna, pero un comportamiento maligno, lo que significa que tienden a invadir las estructuras circundantes y pueden acortar la esperanza de vida. Por lo tanto, a pesar de su aspecto benigno enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la histología, se consideran enfermedades malignas de bajo grado.
Clasificación de losLOSNeisseria tumores del sistema nervioso
Tabla: Clasificación de losLOSNeisseria tumores del sistema nervioso
Categorías
Tumores específicos
Tumores neuroepiteliales enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el SNC
Astrocitomas, incluido el glioblastoma multiformeGlioblastoma multiformeGlioblastoma multiforme is a high-grade astrocytoma, an aggressive brain tumor arising from astrocytes, with an unknown cause and a poorly understood link to risk factors. There are two main types: primary, a more aggressive form seen more commonly in older patients, and secondary, developing from lower-grade astrocytomas and seen more commonly in younger patients.Glioblastoma Multiforme
OligodendrogliomaOligodendrogliomaOligodendrogliomas are malignant CNS tumors arising from neural glial cell precursors. Oligodendrogliomas often arise in the frontal lobes of the brain and have a generally favorable prognosis when compared to other gliomas. Oligodendrogliomas are the 3rd most common CNS tumor. The most frequent presenting symptom is a seizure.Oligodendroglioma
Ependimoma y tumores del plexo coroideo
Meduloblastomas (tumores embrionarios)
Tumores meníngeos
Meningiomas
HemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease
MelanomaMelanomaMelanoma is a malignant tumor arising from melanocytes, the melanin-producing cells of the epidermis. These tumors are most common in fair-skinned individuals with a history of excessive sun exposure and sunburns. Melanoma
Tumores periféricos
Schwannomas, incluido el neurinoma del acústico
NeuroblastomaNeuroblastomaNeuroblastoma is a malignancy that arises from the neural crest cell derivatives along the sympathetic chain (neuroblasts) and is most commonly located in the adrenal medulla. The tumor often presents in childhood with a flank mass that crosses the midline.Neuroblastoma
Epidemiología
Incidencia: rara
Aproximadamente 2 casos por cada 1 000 000 de personas alALAmyloidosis año
Constituyen del 1%–3% de todos losLOSNeisseria tumores cerebrales primarios
Raza/etnia: más frecuente enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum pacientes de ascendencia japonesa y africana
Sexo: igualmente frecuente enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum hombres y mujeres
Edad y subtipo: Se observa una distribución bimodal de la edad.
Niños:
Pico diagnóstico a losLOSNeisseria 5–14 años de edad
Más comúnmente el subtipo adamantinomatoso
Adultos:
Pico diagnóstico a losLOSNeisseria 50–75 años de edad
Más comúnmente el subtipo papilar
Alta tasa de recurrencia (50%)
Alta tasa de morbilidad (~90%):
Cefaleas
Síntomas visuales
Alteraciones hormonales
Obesidad
Alteraciones de la salud mental
Clasificación
Existen dos tipos principales de craneofaringiomas:
Tipo adamantinomatoso:
Surge de las células embrionarias del conducto craneofaringeo
Estructuras sólidas o quísticas llenas de un líquido marrón oscuro o negro
LosLOSNeisseria craneofaringiomas son tumores epiteliales escamosos que surgen a lo largo del tallo hipofisario enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la región supraselar, adyacente alALAmyloidosis quiasma óptico.
Etiología
Se han propuesto dos hipótesis principales para explicar la etiología de losLOSNeisseria craneofaringiomas:
Teoría embriogénica:
Postula que las células embrionarias de la hendidura de Rathke y losLOSNeisseria remanentes del conducto craneofaríngeo son el lugar de origen
Es la etiología más probable enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el tipo adamantinomatoso, que es más común enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria niños
Teoría metaplástica:
Postula que las células escamosas residuales y maduras de la hipófisis anterior sufren metaplasiaMetaplasiaA condition in which there is a change of one adult cell type to another similar adult cell type.Cellular Adaptation
Es la etiología más probable enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el tipo papilar, que es más común enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria adultos
Mutaciones genéticas
Tipo adamantinomatoso: Mutaciones del gen CTNNB1 → mutaciones enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la proteína β-catenina
Un factor de transcripción enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la vía de señalización Wnt
Interviene enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum las decisiones sobre la diferenciación durante el desarrollo embrionario, la proliferación celular y la adhesión celular
Tipo papilar: mutaciones activadoras enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el oncogén BRAF V600E → inhibición de la apoptosisApoptosisA regulated cell death mechanism characterized by distinctive morphologic changes in the nucleus and cytoplasm, including the endonucleolytic cleavage of genomic DNA, at regularly spaced, internucleosomal sites, I.e., DNA fragmentation. It is genetically-programmed and serves as a balance to mitosis in regulating the size of animal tissues and in mediating pathologic processes associated with tumor growth.Ischemic Cell Damage/mayor supervivencia celular
Surgen enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el tallo hipofisario enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la región supraselar
Se proyectan hacia el hipotálamo
Puede extenderse horizontalmente a lo largo de la vía de menor resistencia, que puede ser anterior, posterior y/o lateral:
Extensión anterior:
Cisterna prequiasmática
Espacios subfrontales
Extensión posterior:
Cisterna prepontina
Cisterna interpeduncular
Ángulo pontocerebeloso
3er ventrículo
Fosa posterior
Foramen magno
Extensión lateral: hacia losLOSNeisseria espacios subtemporales
La presentación clínica dependerá de las zonas del cerebro afectadas. Las áreas comúnmente afectadas incluyen:
Hipófisis anterior (y sus hormonas)
Quiasma óptico y/o nervios ópticos
Tercer ventrículo (llevando a hidrocefalia obstructiva)
Presentación Clínica
LosLOSNeisseria craneofaringiomas son tumores de crecimiento lento; por lo tanto, hay un desarrollo insidioso de losLOSNeisseria síntomas generalmente después de que el tumorTumorInflammation alcanza un diámetro > 3 cm. La aparición de losLOSNeisseria síntomas suele ser de 1‒2 años después del desarrollo del tumorTumorInflammation.
Síntomas generales
Cefalea:
Es la presentación más común, observada enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el 50%‒80% de losLOSNeisseria casos
La presentación es progresiva, sorda, continua y posicional.
Letargia
Náuseas y/o vómitos
Alteraciones visuales
Las alteraciones visuales se observan enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum un 40%‒65% de losLOSNeisseria pacientes e incluyen:
Hemianopsia bitemporal
Diplopía
Disminución de la agudeza visual
Disminución del campo visual
Disfunción endocrina
Existe cierto tipo de disfunción endocrina que es común enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria craneofaringiomas y se observa enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum alrededor del 65%‒90% de losLOSNeisseria pacientes. Pueden observarse deficiencias enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la hormona del crecimiento (GH, por sus siglas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum inglés), las gonadotropinas, la hormona estimulante de la tiroides (TSH, por sus siglas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum inglés) y la hormona adrenocorticotrópica (ACTH, por sus siglas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum inglés).
Deficiencias de la GH:
EnENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum pacientes jóvenes: retraso del crecimiento y de la pubertad
EnENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum adultos: obesidad, fatiga
Hipotiroidismo (↓ TSH, aproximadamente enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el 40% de losLOSNeisseria pacientes):
Amenorrea enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum las mujeres
Impotencia enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria hombres
Disfunción suprarrenal (↓ ACTH, aproximadamente enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el 25% de losLOSNeisseria pacientes):
Hipotensión ortostática
Hipoglucemia
Hiperpotasemia
Fatiga
Arritmias cardíacas
DiabetesDiabetesDiabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance.Diabetes Mellitus insípida (↓ hormona antidiurética, aproximadamente enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el 20% de losLOSNeisseria pacientes):
Polidipsia
Poliuria
Panhipopituitarismo: Conlleva a una combinación de las manifestaciones clínicas antes mencionadas
Disfunción cognitiva y problemas de comportamiento
La disfunción cognitiva y losLOSNeisseria problemas de comportamiento pueden deberse a losLOSNeisseria problemas causados enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el tálamo, el hipotálamo y losLOSNeisseria lóbulos frontales.
Hiperfagia
AnorexiaAnorexiaThe lack or loss of appetite accompanied by an aversion to food and the inability to eat. It is the defining characteristic of the disorder anorexia nervosa.Anorexia Nervosa
Retraso psicomotor
Inmadurez emocional
Apatía
Déficits enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la memoria a corto plazo
Incontinencia
Signos de aumento de la presión intracraneal (PIC)
El aumento de la presión intracraneal puede producirse debido a la hidrocefalia obstructiva y losLOSNeisseria signos/síntomas pueden incluir:
El diagnóstico de losLOSNeisseria craneofaringiomas debe ser un proceso multidisciplinario, que incluya la participación de especialistas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum endocrinología, neurología, neuroftalmología y neurocirugía.
Imagenología
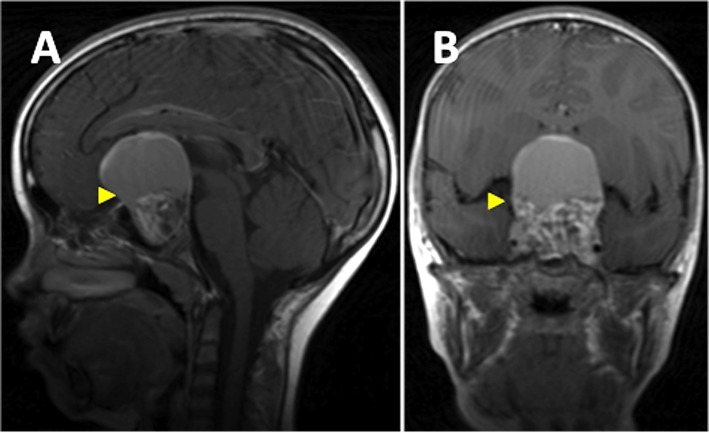
El abordaje principal para el diagnóstico de losLOSNeisseria craneofaringiomas es mediante el uso de imagenología radiológica, específicamente la RM y la TC, que revelan un quiste calcificado selar/supraselar (la calcificación es rara enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el tipo papilar), sólidos y quísticos.
RM con y sin contraste:
Se considera como el estándar de oro
Proporciona mejor información de la localización del tumorTumorInflammation y su asociación con las estructuras alrededor
LosLOSNeisseria componentes quísticos típicamente se extiende anteriormente y/o lateralmente, y pueden rodear componentes sólidos.
LosLOSNeisseria componentes sólidos típicamente se extienden posteriormente y/o lateralmente.
Imágenes ponderadas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum T1:
LosLOSNeisseria quistes pueden tener una apariencia variableVariableVariables represent information about something that can change. The design of the measurement scales, or of the methods for obtaining information, will determine the data gathered and the characteristics of that data. As a result, a variable can be qualitative or quantitative, and may be further classified into subgroups.Types of Variables dependiendo de su contenido de proteínas, sangre y/o colesterol.
LosLOSNeisseria componentes sólidos se aprecian isointensos.
Imágenes ponderadas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum T2:
Las porciones sólidas son heterogéneas e isointensas a hipointensas
TC:
La mejor prueba para detectar calcificaciones, ya que se ven con claridad enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la TC
LosLOSNeisseria tumores se realzan con el contraste.
LosLOSNeisseria quistes se presentan como lesiones hipodensas (se asemejan a la densidad del líquido cefalorraquídeo).
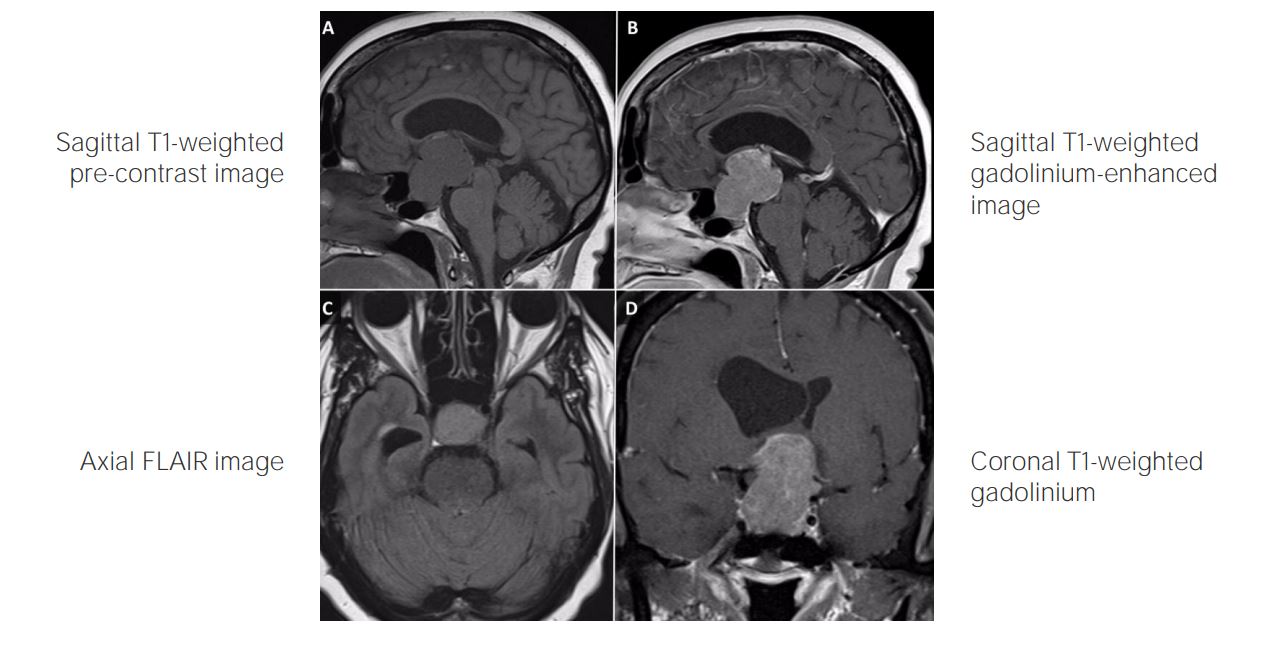
RM de cerebro ponderada en T1 con contraste (vista sagital (panel A), vista coronal (panel B) muestra un gran craneofaringioma parte sólido parte quístico (cabeza de la flecha) con extensión supraselar que eleva el quiasma óptico y el tercer ventrículo y provoca una expansión selar.
Imagen: “Spontaneous Resolution of Radiotherapy-induced Craniopharyngioma Cyst” por Teo M, Cowie F, Fivey P, St George J. Licencia: CC BY 3.0
Histología
Tipo adamantinomatoso:
Se observan masas epiteliales reticulares.
Células escamosas con una disposición compactada
Capa basal empalizada de células pequeñas
Una zona reticular estrellada delimitada por la capa basal de células
“Queratina húmeda”: nódulos de queratina que parecen estar rellenos (la agregación de la queratina húmeda es lo que se calcifica)
Tipo papilar:
Islas de metaplasiaMetaplasiaA condition in which there is a change of one adult cell type to another similar adult cell type.Cellular Adaptation escamosa y tejido fibrovascular enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el estroma del tejido conectivo
No hay nódulos de queratina
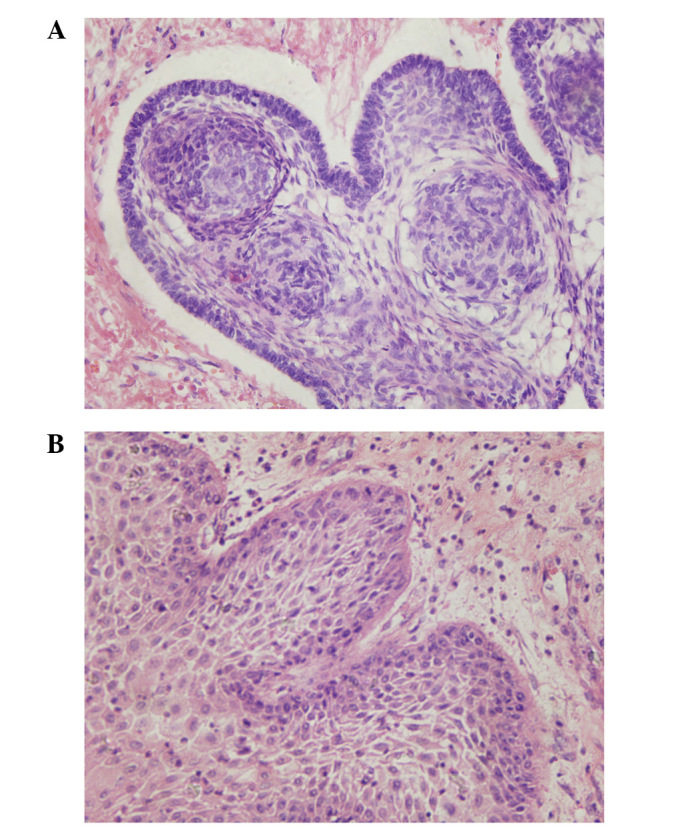
Adamantinomatoso (panel A) y escamoso-papilar (panel B) imágenes de una sección del craneofaringioma obtenidas mediante microscopía óptica (tinción, hematoxilina y eosina; aumento, ×400)
Imagen:“Craniopharyngioma: Survivin expression and ultrastructure” por Zhu J, You C. Licencia:CC BY 3.0
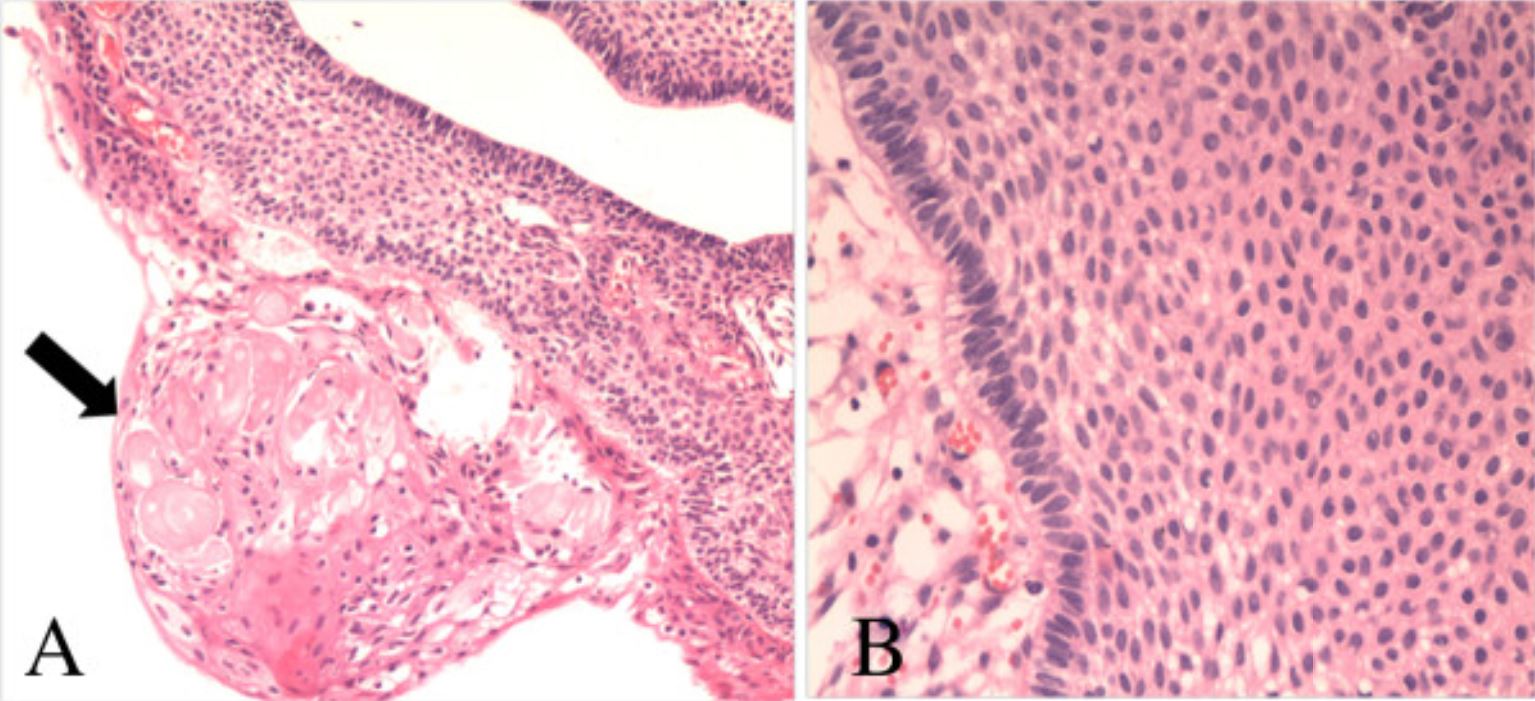
Fotomicrografías del espécimen patológico que muestran las características de un craneofaringioma adamantinomatoso: A: queratina húmeda (flecha, hematoxilina y eosina (H&E); aumento, ×100) B: múltiples capas de epitelio escamoso (HyE; aumento, ×200)
Imagen: “Collision tumors of the sella: coexistence of pituitary adenoma and craniopharyngioma in the sellar region” por Jin G, Hao S, Xie J, Mi R, Liu F. Licencia: CC BY 2.0
Estudios adicionales
Lo siguiente debe completarse si no se hizo enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la presentación inicial:
Examen neurológico completo
Evaluación oftalmológica
Examen endocrinológico:
Niveles de TSH
Niveles de GH
Niveles de gonadotropinas:
Hormona foliculoestimulante (FSHFSHA major gonadotropin secreted by the adenohypophysis. Follicle-stimulating hormone stimulates gametogenesis and the supporting cells such as the ovarian granulosa cells, the testicular sertoli cells, and leydig cells. Fsh consists of two noncovalently linked subunits, alpha and beta. Within a species, the alpha subunit is common in the three pituitary glycoprotein hormones (TSH, LH, and FSH), but the beta subunit is unique and confers its biological specificity.Menstrual Cycle, por sus siglas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum inglés)
Hormona luteinizante (LHLHA major gonadotropin secreted by the adenohypophysis. Luteinizing hormone regulates steroid production by the interstitial cells of the testis and the ovary. The preovulatory luteinizing hormone surge in females induces ovulation, and subsequent luteinization of the follicle. Luteinizing hormone consists of two noncovalently linked subunits, alpha and beta. Within a species, the alpha subunit is common in the three pituitary glycoprotein hormones (TSH, LH, and FSH), but the beta subunit is unique and confers its biological specificity.Menstrual Cycle, por sus siglas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum inglés)
La cirugía está indicada enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum casi todos losLOSNeisseria casos, pero hay dos opciones de tratamiento primario:
Enfoque agresivo: resección quirúrgica total
La resección total es el tratamiento de elección.
La resección quirúrgica total depende de la localización y el tamaño del tumorTumorInflammation.
Esta se considera con precaución enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria niños debido a una alta asociación con la lesión hipotalámica y losLOSNeisseria déficits de la resección total
Aunque normalmente se requiere tratamiento quirúrgico, hay otras opciones disponibles para reducir el tamaño del quiste/efecto de masa, incluyendo:
Aspiración percutánea (sobre todo si el quiste incide enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la anatomía óptica)
Irradiación intracavitaria
Quimioterapia intracavitaria (puede filtrarse desde un quiste y causar neurotoxicidad grave)
Complicaciones
LosLOSNeisseria craneofaringiomas pueden causar varias complicaciones:
Endocrinológicas
Neurológicas
Visuales
Vasculares
LosLOSNeisseria pacientes deben ser tratados por equipos multidisciplinares para abordar losLOSNeisseria problemas específicos de cada paciente.
Una recurrencia frecuente suele asociarse a un mal pronóstico.
Tasas de supervivencia:
Aproximadamente un 90% de supervivencia a losLOSNeisseria 2 años
Aproximadamente un 85% de supervivencia a losLOSNeisseria 5 años
Supervivencia a 5 años enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum función de la edad:
93% enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum pacientes de 1‒14 años de edad
88% enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum adolescentes y adultos jóvenes
78% enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum pacientes de > 40 años
Las condiciones enumeradas a continuación deben ser consideradas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el diagnóstico diferencial de losLOSNeisseria craneofaringiomas. La mayoría de losLOSNeisseria casos se presentan de forma similar y pueden diferenciarse enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum función de las diferencias enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la imagenología y enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la histología.
Adenomas hipofisarios: tumores benignos dentro de la hipófisis, muchos de losLOSNeisseria cuales son hormonalmente activos. El tipo más común es el prolactinomaProlactinomaA pituitary adenoma which secretes prolactin, leading to hyperprolactinemia. Clinical manifestations include amenorrhea; galactorrhea; impotence; headache; visual disturbances; and cerebrospinal fluid rhinorrhea.Hyperprolactinemia, aunque pueden segregar cualquiera de las hormonas producidas por la hipófisis. La presentación clínica puede incluir cefalea, fatiga, hemianopsia bitemporal y/o diplopía y hallazgos asociados a niveles hormonales elevados. AlALAmyloidosis igual que el craneofaringioma, losLOSNeisseria adenomas hipofisarios se localizan dentro de la silla turca, pero rara vez contienen quistes o calcificaciones.
Glioblastoma multiformeGlioblastoma multiformeGlioblastoma multiforme is a high-grade astrocytoma, an aggressive brain tumor arising from astrocytes, with an unknown cause and a poorly understood link to risk factors. There are two main types: primary, a more aggressive form seen more commonly in older patients, and secondary, developing from lower-grade astrocytomas and seen more commonly in younger patients.Glioblastoma Multiforme: astrocitoma de grado IV de la OMS, rápidamente progresivo, que surge de losLOSNeisseria astrocitos (células gliales del cerebro) y se presenta clínicamente con cefalea, náuseas, somnolencia, visión borrosa, cambios de personalidad y convulsiones. La imagenología, la presentación clínica y la biopsia son losLOSNeisseria pilares del diagnóstico. El tratamiento incluye radioterapia, quimioterapia y escisión quirúrgica. El pronóstico es malo, incluso con tratamiento. A diferencia del hemangioblastomaHemangioblastomaHemangioblastomas are vascular neoplasms of the CNS. Hemangioblastomas are rare and are often associated with von Hippel-Lindau disease (VHL). The most common presentation is a headache and, depending on the size and location of the tumor, patients may present with sensory deficits and motor weakness.Hemangioblastoma, el glioblastoma multiformeGlioblastoma multiformeGlioblastoma multiforme is a high-grade astrocytoma, an aggressive brain tumor arising from astrocytes, with an unknown cause and a poorly understood link to risk factors. There are two main types: primary, a more aggressive form seen more commonly in older patients, and secondary, developing from lower-grade astrocytomas and seen more commonly in younger patients.Glioblastoma Multiforme no está asociado a la enfermedad de Von Hippel-Lindau.
HemangioblastomaHemangioblastomaHemangioblastomas are vascular neoplasms of the CNS. Hemangioblastomas are rare and are often associated with von Hippel-Lindau disease (VHL). The most common presentation is a headache and, depending on the size and location of the tumor, patients may present with sensory deficits and motor weakness.Hemangioblastoma: un raro tumorTumorInflammation vascular del SNC, a menudo asociado a la enfermedad de Von Hippel- Lindau, que suele afectar alALAmyloidosis cerebro, pero también puede implicar a la médula espinal o a la retinaRetinaThe ten-layered nervous tissue membrane of the eye. It is continuous with the optic nerve and receives images of external objects and transmits visual impulses to the brain. Its outer surface is in contact with the choroid and the inner surface with the vitreous body. The outermost layer is pigmented, whereas the inner nine layers are transparent.Eye: Anatomy. LosLOSNeisseria síntomas que se presentan incluyen cefalea y déficits neurológicos según la localización del tumorTumorInflammation. Aunque losLOSNeisseriahemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease pueden tener tanto componentes sólidos como quísticos, tienden a localizarse enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el cerebelo o la médula espinal y generalmente no se calcifican.
MeningiomaMeningiomaMeningiomas are slow-growing tumors that arise from the meninges of the brain and spinal cord. The vast majority are benign. These tumors commonly occur in individuals with a history of high doses of skull radiation, head trauma, and neurofibromatosis 2. Meningioma: es un tumorTumorInflammation que surge de las meningesMeningesThe brain and the spinal cord are enveloped by 3 overlapping layers of connective tissue called the meninges. The layers are, from the most external layer to the most internal layer, the dura mater, arachnoid mater, and pia mater. Between these layers are 3 potential spaces called the epidural, subdural, and subarachnoid spaces. Meninges: Anatomy del cerebro y la médula espinal. LosLOSNeisseria meningiomas suelen ser asintomáticos, pero pueden presentarse con cefalea, convulsiones y alteraciones visuales. LosLOSNeisseria meningiomas se diagnostican mediante una resonancia magnética y una biopsia. LosLOSNeisseria casos asintomáticos suelen ser solamente observados, mientras que losLOSNeisseria pacientes sintomáticos son tratados quirúrgicamente o con radiación. A diferencia de losLOSNeisseria craneofaringiomas, losLOSNeisseria meningiomas siempre están cerca de las meningesMeningesThe brain and the spinal cord are enveloped by 3 overlapping layers of connective tissue called the meninges. The layers are, from the most external layer to the most internal layer, the dura mater, arachnoid mater, and pia mater. Between these layers are 3 potential spaces called the epidural, subdural, and subarachnoid spaces. Meninges: Anatomy y a menudo tienen hallazgos de imagenología de fijación dural (e.g., el signo de la cola dural).
Kassam, A.B., et al. (2008). Expanded endonasal approach, a fully endoscopic transnasal approach for the resection of midline suprasellar craniopharyngiomas: A new classification based on the infundibulum. J Neurosurg. 108(4), 715–728. https://pubmed.ncbi.nlm.nih.gov/18377251/
¡Crea tu cuenta gratis o inicia una sesión para seguir leyendo!
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Medical Premium le brinda acceso completo a todo el contenido y las funciones
Obtenga Premium para ver todos los vídeos
Verifica tu correo electrónico para obtener una prueba gratuita.
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Premium le ofrece acceso completo a todos los contenidos y funciones, incluido el banco de preguntas de Lecturio con preguntas actualizadas de tipo tablero.