La anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types aplásica es una afección rara y potencialmente mortal caracterizada por pancitopenia e hipocelularidad de la médula ósea ( en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum ausencia de células anormales) que refleja daño a las células madre hematopoyéticas. La anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types aplásica puede ser adquirida o heredada, sin embargo, la mayoría de los LOS Neisseria casos de anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types aplásica son adquiridos y causados por daño autoinmune a las células madre hematopoyéticas. Las causas y asociaciones adquiridas específicamente conocidas de anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types aplásica incluyen medicamentos, productos químicos, altas dosis de radiación en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum todo el cuerpo, infecciones virales, enfermedades inmunitarias y embarazo. Los LOS Neisseria síndromes hereditarios o constitucionales asociados con anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types aplásica incluyen anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types de Fanconi, disqueratosis congénita y síndrome de Down. Las terapias incluyen soporte transfusional, inmunosupresión y trasplante de médula ósea.
Last updated: Dec 15, 2025
Condiciones heredadas:
Anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types aplasica adquirida:
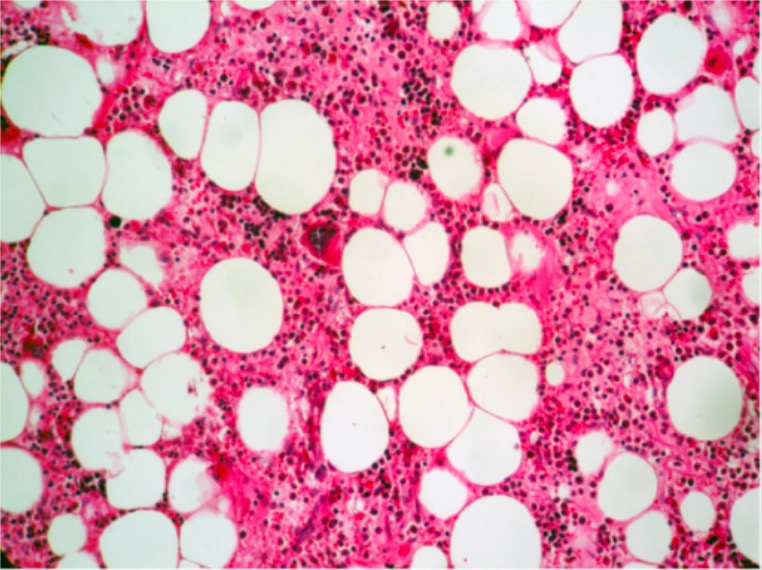
Hematopoyesis normal (las 3 líneas celulares presentes) con un megacariocito cerca del centro del campo (H&E, 200x)
Imagen: “Histologic findings” por Department of Pathology, Ibn Sina University Hospital, Rabat, Morocco. Licencia: CC BY 2.0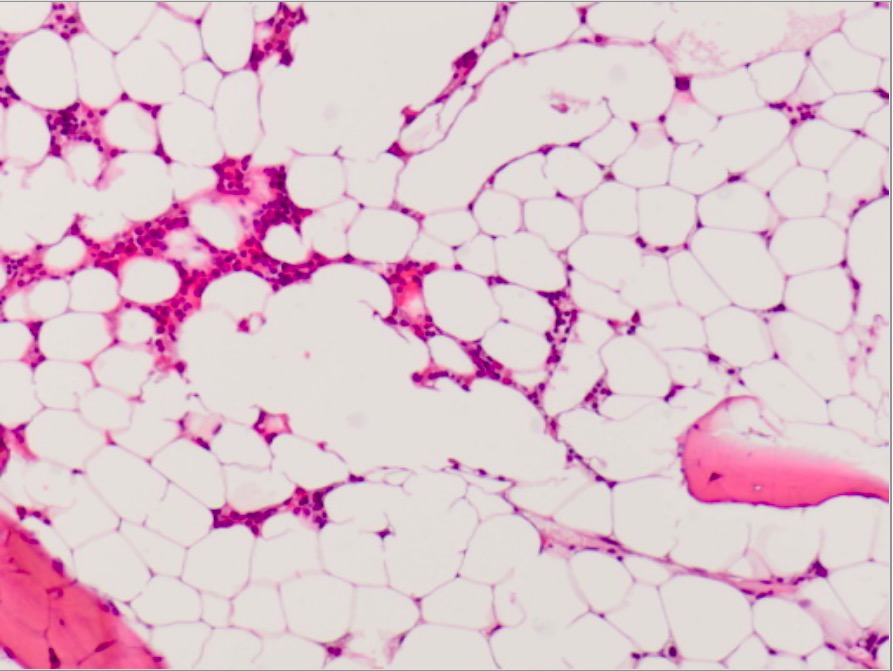
Fotografía microscópica de médula ósea en anemia aplásica:
Médula ósea marcadamente hipocelular de un paciente con anemia aplásica adquirida (que muestra solo una hematopoyesis escasa y en su mayoría células grasas)
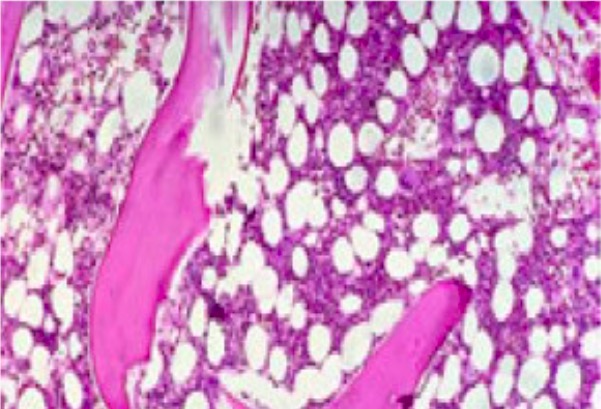
Médula ósea con aproximadamente 50% de celularidad (consistente con la médula ósea de una persona sana de 50 años)
Imagen: “Mild hypoblastic marrow” por Hasnaa Aboelwafa et al. Licencia: CC BY 2.0, recortada por Lecturio.Pancitopenia con médula ósea hipocelular que muestra: