


A doença de Wilson (degeneração hepatolenticular) é uma doença autossómica recessiva causada por várias mutações no gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics ATP7B, que regula o transporte de cobre dentro dos hepatócitos. A disfunção desse mecanismo de transporte leva à acumulação de quantidades anormais de cobre no fígado, cérebro, olhos e outros órgãos, com consequentes distúrbios hepáticos, neurológicos e psiquiátricos, de expressão variável. O envolvimento do fígado pode manifestar-se como hepatite, insuficiência hepática ou cirrose, enquanto o envolvimento dos gânglios da base causa os sinais extrapiramidais. A maioria dos pacientes é diagnosticada entre as idades de 5 e 35 anos (média: 13 anos). O diagnóstico é estabelecido se o paciente tiver a ceruloplasmina plasmática baixa, depósitos de cobre na córnea (anéis de Kayser-Fleischer) e níveis elevados de cobre na urina. No entanto, muitas vezes são necessários outros exames, pois nem todos os pacientes terão todos estes achados. O prognóstico é bom para pacientes sem doença hepática avançada e que são tratados com os agentes quelantes penicilamina ou trientina. A doença de Wilson não tratada é fatal, podendo os pacientes morrer de cirrose, insuficiência hepática aguda ou complicações devido à doença neurológica progressiva.
Last updated: Jan 7, 2026
A doença de Wilson geralmente apresenta-se em crianças e adultos jovens. Raramente se manifesta após os 40 ano. As manifestações clínicas são principalmente hepáticas, neurológicas e psiquiátricas e podem incluir:
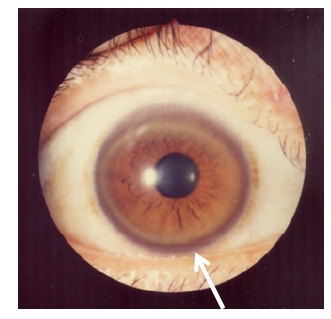
Anel de Kayser-Fleischer: deposição de cobre na córnea
Imagem : Kayser-Fleischer rings” por Bentham Science Publishers, 2012. Licença: CC-BY-2.5Apresentação clínica da doença de Wilson: ABCD
