La talasemia es una causa hereditaria de anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types microcítica hipocrómica y es el resultado de una deficiencia en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum las cadenas de globina α o β, lo que provoca una hemoglobinopatía. La presentación de la talasemia depende del número de cadenas defectuosas presentes y puede variar desde ser asintomática hasta provocar que los LOS Neisseria pacientes más gravemente afectados sean dependientes de transfusiones. El diagnóstico puede confirmarse mediante una electroforesis de hemoglobina, que revelará la presencia de cadenas de α o β-globina anormales.
Last updated: Dec 15, 2025
| Número de genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure eliminados/genotipo | Enfermedad | Resultado |
|---|---|---|
| 1 (αα/α-) |
α-Talasemia mínima | Portador silencioso |
| 2 (α-/α-; trans) (αα/–; cis CIS Multiple Sclerosis) |
α-Talasemia menor |
|
| 3 (α-/–) |
Hemoglobina H (4 cadenas β) | Anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types moderada a severa |
| 4 (–/–) |
Hemoglobina Barts (4 cadenas γ) | Hidropesía fetal (incompatible con la vida) |
| Número de genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure eliminados/genotipo | Enfermedad | Resultado |
|---|---|---|
| β0/β o β+/β | Talasemia menor | Asintomático (leve) |
| β0/β0 o β+/β+ | Talasemia mayor ( anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types de Cooley) | Dependiente de transfusiones |
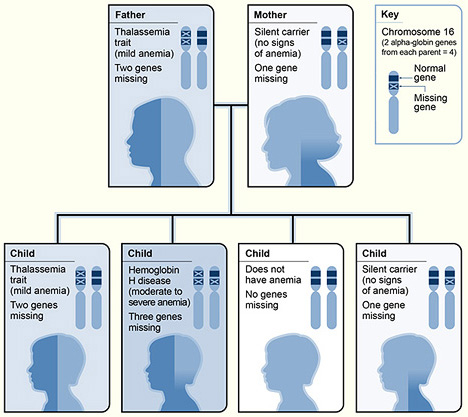
Un ejemplo de herencia de la α-talasemia
Imagen: “The picture shows one example of how alpha thalassemia is inherited” por National Heart Lung and Blood Institute (NIH). Licencia: Dominio Público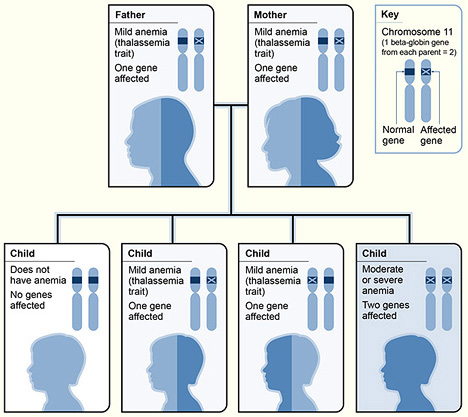
Un ejemplo de herencia de la β-talasemia
Imagen: “The picture shows one example of how beta thalassemia is inherited” por National Heart Lung and Blood Institute (NIH). Licencia: Dominio Público| Enfermedad | Presentación |
|---|---|
| α-Talasemia mínima | Asintomática |
| α-Talasemia menor |
|
| Enfermedad hemoglobina H |
|
| Hemoglobina Barts |
|
| β-Talasemia menor |
|
| β-Talasemia mayor ( anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types de Cooley) |
|
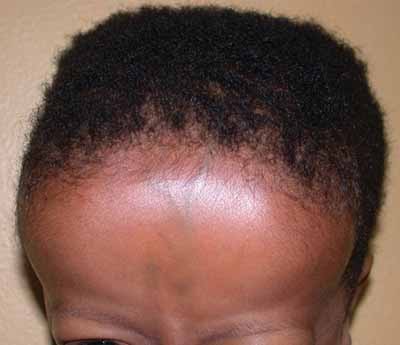
La prominencia frontal se debe a una necesidad excesiva de hematopoyesis
Imagen: “Frontal bossing (abnormally enlarged forehead) in a child” por National Human Genome Research Institute (NIH). Licencia: Dominio Público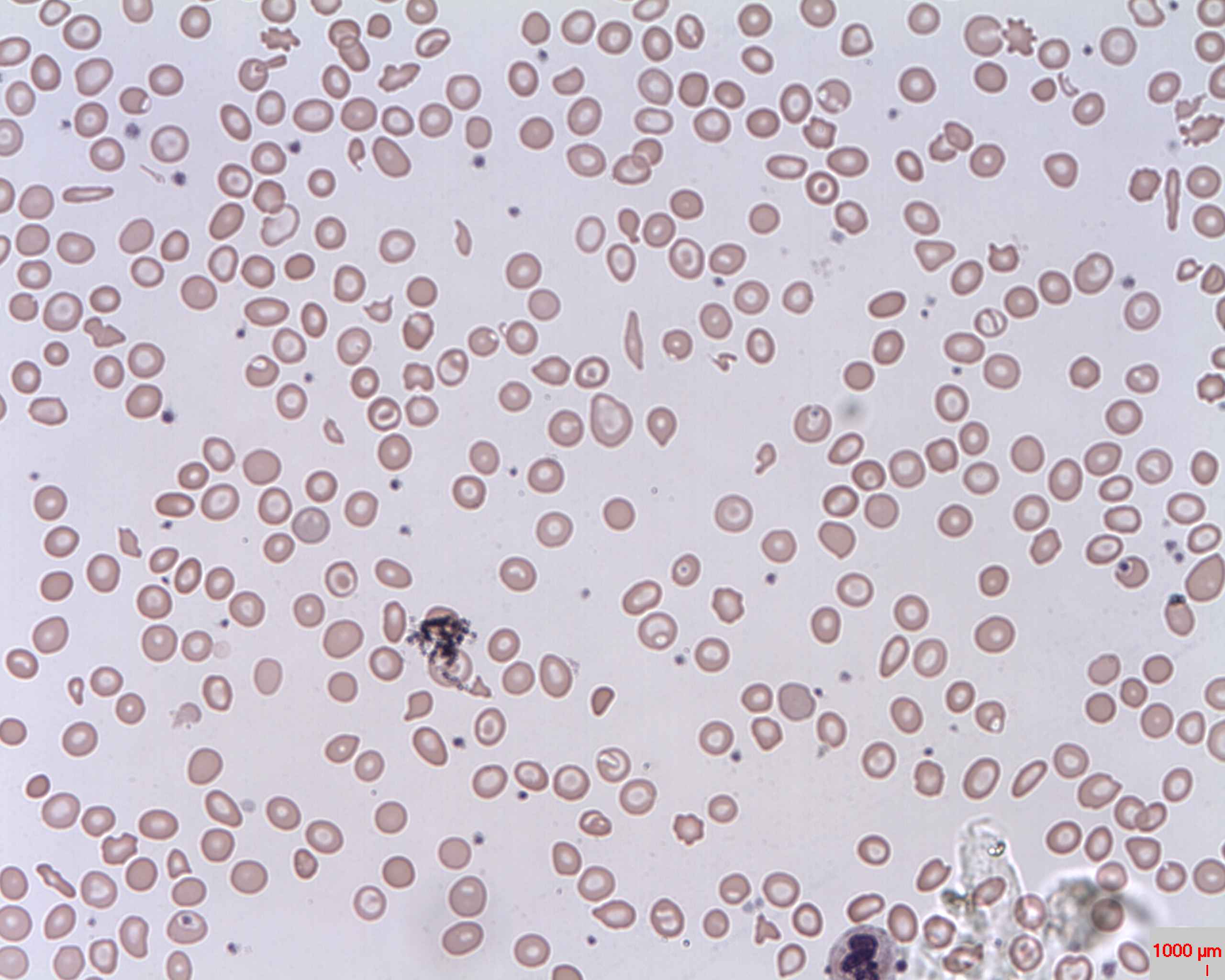
Frotis periférico: anemia microcítica hipocrómica con codocitos (ojo de buey); también se observa anisocitosis (eritrocitos de tamaño desigual)
Imagen: “Target cells (Codocytes, Leptocytes or Mexican hat cells)” por Prof. Osaro Erhabor. Licencia: CC0 1.0El diagnóstico diferencial incluye otras afecciones asociadas a la anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types microcítica o a codocitos.