El síndrome de Marfan es una enfermedad genética de herencia autosómica dominante. El síndrome de Marfan afecta la elasticidad de los LOS Neisseria tejidos conectivos de todo el cuerpo, sobre todo en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria sistemas cardiovascular, ocular y musculoesquelético. También se ven afectados la piel, los LOS Neisseria pulmones y el sistema nervioso central. Los LOS Neisseria pacientes suelen ser altos, con extremidades y dedos de las manos y de los LOS Neisseria pies largos, y articulaciones hipermóviles. Las afecciones asociadas incluyen el aneurisma o la disección de la aorta Aorta The main trunk of the systemic arteries. Mediastinum and Great Vessels: Anatomy, el prolapso de la válvula mitral y la luxación del cristalino. El diagnóstico se realiza clínicamente con criterios establecidos, y las pruebas genéticas se realizan solamente cuando pueden influir en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el tratamiento. El tratamiento médico o quirúrgico se basa en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum las manifestaciones clínicas. El compromiso cardiovascular requiere seguimiento estrecho, ya que es la principal causa de mortalidad.
Last updated: Dec 15, 2025

Familiares con síndrome de Marfan
Imagen: “Family members with Marfan’s syndrome” por Birjand Atherosclerosis and Coronary Artery Research Centre, Birjand University of Medical Sciences, Birjand, Iran. Licencia: CC BY 3.0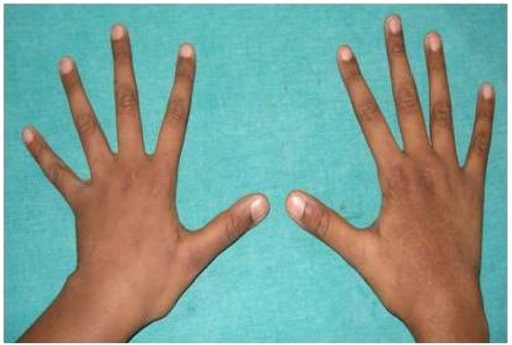
Fotografía que muestra aracnodactilia
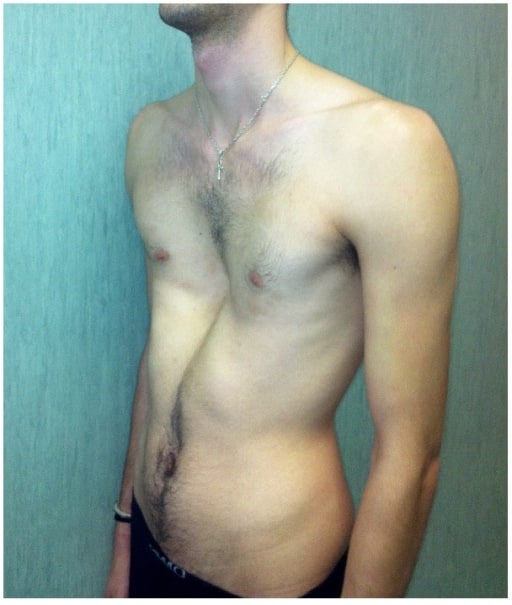
Pectus excavatum grave en un hombre afectado por el síndrome de Marfan
Imagen: “fig2” por Fernando De Maio et al. Licencia: CC BY 4.0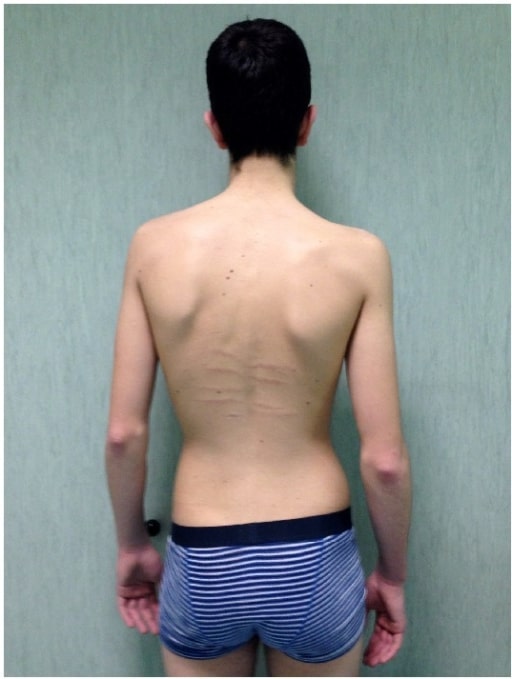
Un hombre afectado por el síndrome de Marfan con escoliosis, además de estrías cutáneas dorsales y reducción de la extensión del codo
Imagen: “fig5” por Fernando De Maio et al. Licencia: CC BY 4.0
Un paciente con síndrome de Marfan con el signo de la muñeca y del pulgar:
(a) El “signo de la muñeca” es positivo cuando el pulgar se superpone al quinto dedo al agarrar la muñeca contralateral.
(b) El “signo del pulgar” es positivo cuando el pulgar se extiende mucho más allá del borde cubital de la mano al superponer los dedos.
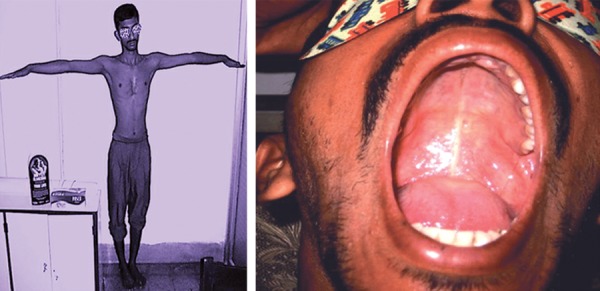
Una envergadura de los brazos superior a la altura del paciente, y un paladar alto y arqueado
Imagen: “Arm span more than height and high arched palate” por Consultant, Eye and Glaucoma Care, Gariahat, Kolkata-700029, West Bengal, India. Licencia: CC BY 3.0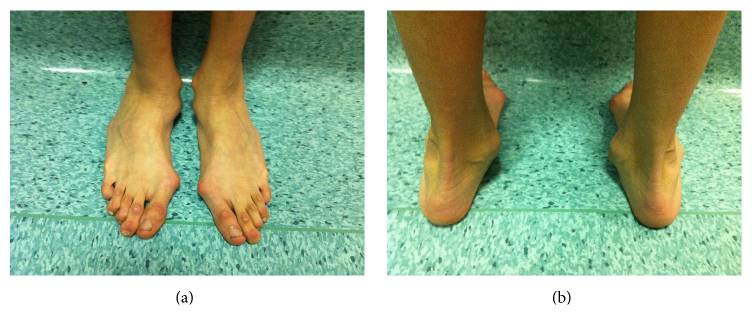
Deformación del retropié con un marcado valgo del talón en un hombre afectado por el síndrome de Marfan
Imagen: “Hindfoot deformity” por Fernando De Maio et al. Licencia: CC BY 4.0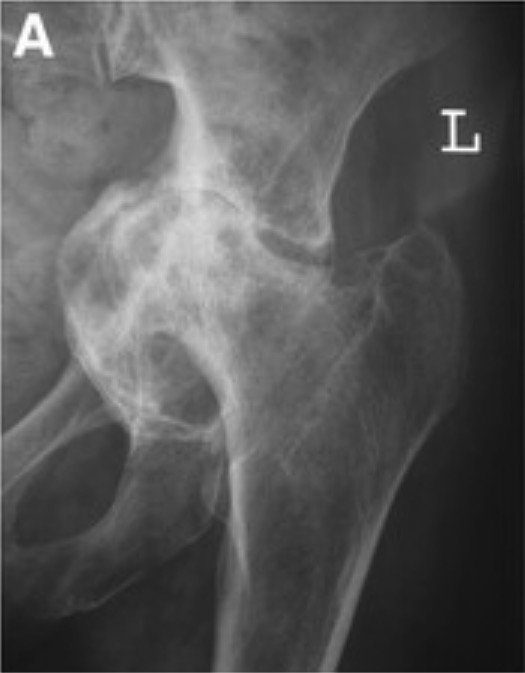
Radiografía que demuestra la protrusión acetabular: Obsérvese que la cabeza del fémur y la cavidad se proyectan hacia la pelvis.
Imagen: “Grade III protrusio acetabuli” por Orthopaedic Research Fellow, Royal Infirmary of Edinburgh, Little France EH16 4SA, UK. Licencia: CC BY 2.0, editada por Lecturio.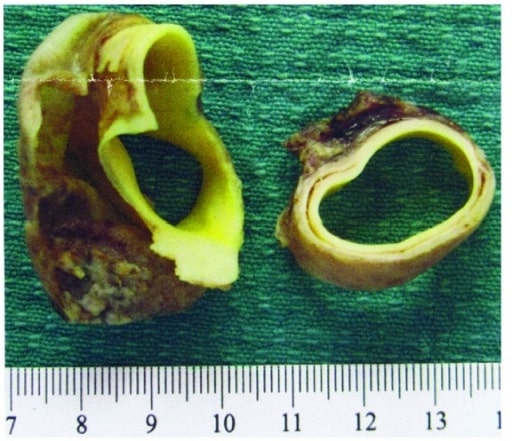
Muestra macroscópica de patología de un colgajo de disección aórtica y de la pared aórtica
Imagen: “Aortic dissection flap and aortic wall” por Department of Obstetrics and Gynaecology, Women’s and Children’s Hospital, Adelaide, SA 5006, Australia. Licencia: CC BY 3.0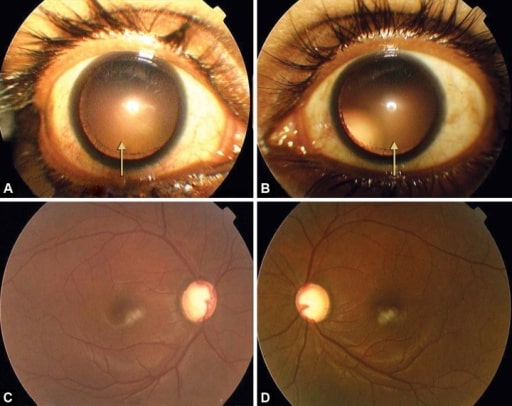
A y B: subluxación temporal y ascendente simétrica del cristalino en la dilatación
C y D: ahuecamientos glaucomatosos avanzados en ambos ojos
| Signo de la muñeca y el pulgar |
|
| Deformación de pectus carinatum Pectus carinatum A developmental anomaly characterized by abnormal anterior protrusion of the sternum and adjacent costal cartilage. Cardiovascular Examination | 2 puntos |
| Pectus excavatum Pectus Excavatum Cardiovascular Examination o asimetría torácica | 1 punto |
| Deformación del retropié | 2 puntos |
| Pie plano | 1 punto |
| Neumotórax | 2 puntos |
| Ectasia dural | 2 puntos |
| Protrusión acetabular | 2 puntos |
| ↓ Relación entre el segmento superior y el segmento inferior, y ↑ relación entre la envergadura de brazos/altura, y sin escoliosis severa. | 1 punto |
| Escoliosis o cifosis | 1 punto |
| ↓ Extensión del codo | 1 punto |
Rasgos faciales (
al
AL
Amyloidosis menos 3 de 5)
|
1 punto |
| Estrías cutáneas | 1 punto |
| Miopía | 1 punto |
| Prolapso de la válvula mitral | 1 punto |
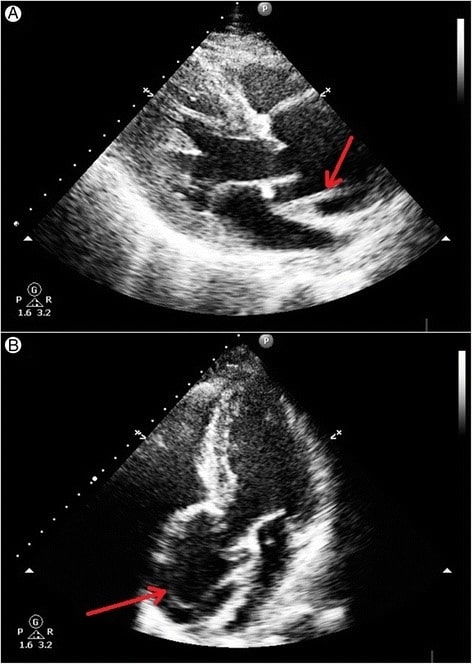
Ultrasonido cardíaco transtorácico que muestra un desgarro de la íntima de la raíz aórtica dilatada por encima del nivel de la válvula aórtica:
A: Se observa un desgarro lineal de la íntima justo por encima de la válvula aórtica en sístole.
B: El colgajo lineal de la capa íntima es visible en la raíz aórtica dilatada.
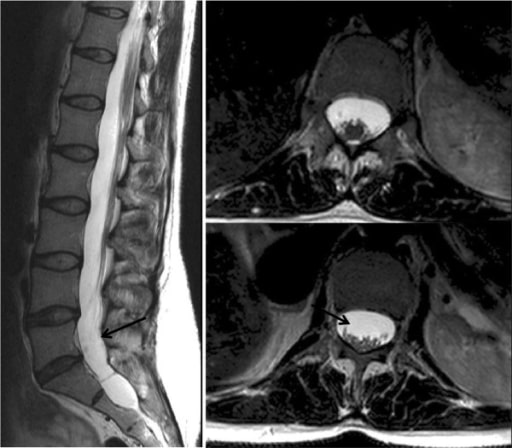
Resonancia magnética que demuestra una ectasia dural: Se trata de un abombamiento o ensanchamiento del saco dural; puede estar asociado a la herniación de los manguitos de las raíces nerviosas a través de sus forámenes asociados (ver flechas).
Imagen: “CMR” por Manchester Heart Centre, Manchester Royal Infirmary, Oxford Road, Manchester M13 9WL, UK. Licencia: CC BY 2.0El tratamiento del SMF se basa en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum las manifestaciones clínicas. Se necesita un equipo multidisciplinario de cardiólogos, oftalmólogos, ortopedistas y cirujanos cardiovasculares.
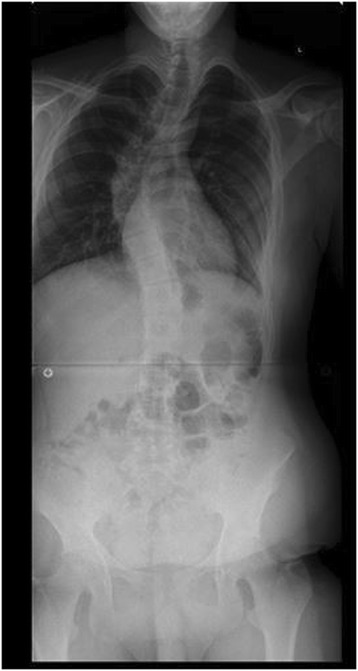
Radiografía preoperatoria de un adolescente con síndrome de Marfan y escoliosis
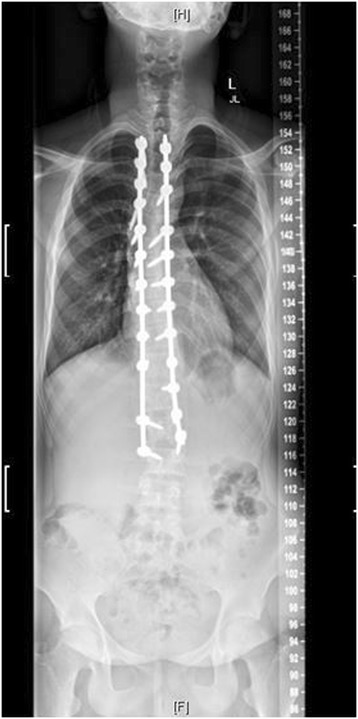
Radiografía postoperatoria del mismo paciente
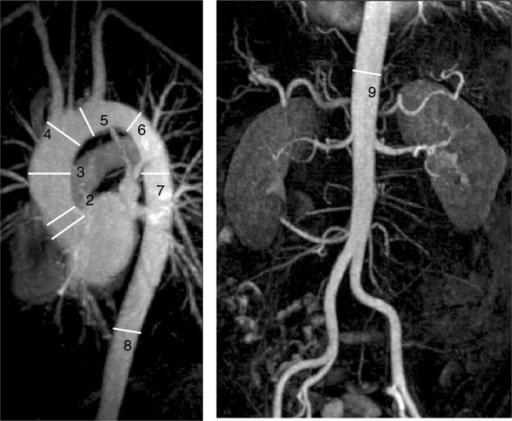
RM cardíaca de la aorta que demuestra los puntos imagenológicos de referencia estándar de la aorta toracoabdominal, utilizados para la monitorización: Esta imagen muestra la dilatación de la aorta ascendente media (3).
Imagen: “F11” por Manchester Heart Centre, Manchester Royal Infirmary, Oxford Road, Manchester M13 9WL, UK. Licencia: CC BY 2.0