Los síndromes de la neoplasia endocrina múltiple son afecciones hereditarias autosómicas dominantes caracterizadas por 2 o más tumores productores de hormonas que afectan a los órganos endocrinos. Existen diferentes tipos de NEM, concretamente NEM1–4. El síndrome de NEM1 está asociado a la mutación del gen MEN1 y tiene una predilección por el hiperparatiroidismo primario, los adenomas hipofisarios y los tumores pancreáticos (las 3 P). Debido a la mutación del protooncogén RET, el síndrome NEM2 puede clasificarse además en NEM2A y NEM2B. El carcinoma medular de tiroides y el feocromocitoma son características comunes. La variante NEM2A se asocia a hiperparatiroidismo primario, mientras que la NEM2B (también considerada NEM3) se asocia a neuromas y hábito marfanoide. La entidad más reciente y rara, NEM4, tiene características de NEM1 pero es el resultado de mutaciones en CDKN1B. El diagnóstico es clínico, y los tumores se detectan basándose en la imagenología y los niveles hormonales relacionados. Las pruebas genéticas desempeñan un papel crucial en los síndromes de NEM2 para determinar el tratamiento posterior. El tratamiento depende de los tumores presentes y de la mutación genética.
Last updated: Dec 15, 2025
Los síndromes de neoplasia endocrina múltiple (NEM) son trastornos genéticos caracterizados por la presencia de ≥ 2 tumores endocrinos.
| NEM1 | NEM2A y NEM2B | NEM4 | |
|---|---|---|---|
| Patrón | Autosómico dominante | Autosómico dominante | Autosómico dominante |
| Mutación genética | Gen MEN1 en el cromosoma 11 (11q13) | Protooncogén RET en el cromosoma 10 (10q11.2) | CDKN1B en el cromosoma 12 (12p13) |
| Características clínicas |
|
NEM2A
NEM2B
|
|
| Tratamiento |
|
|
|
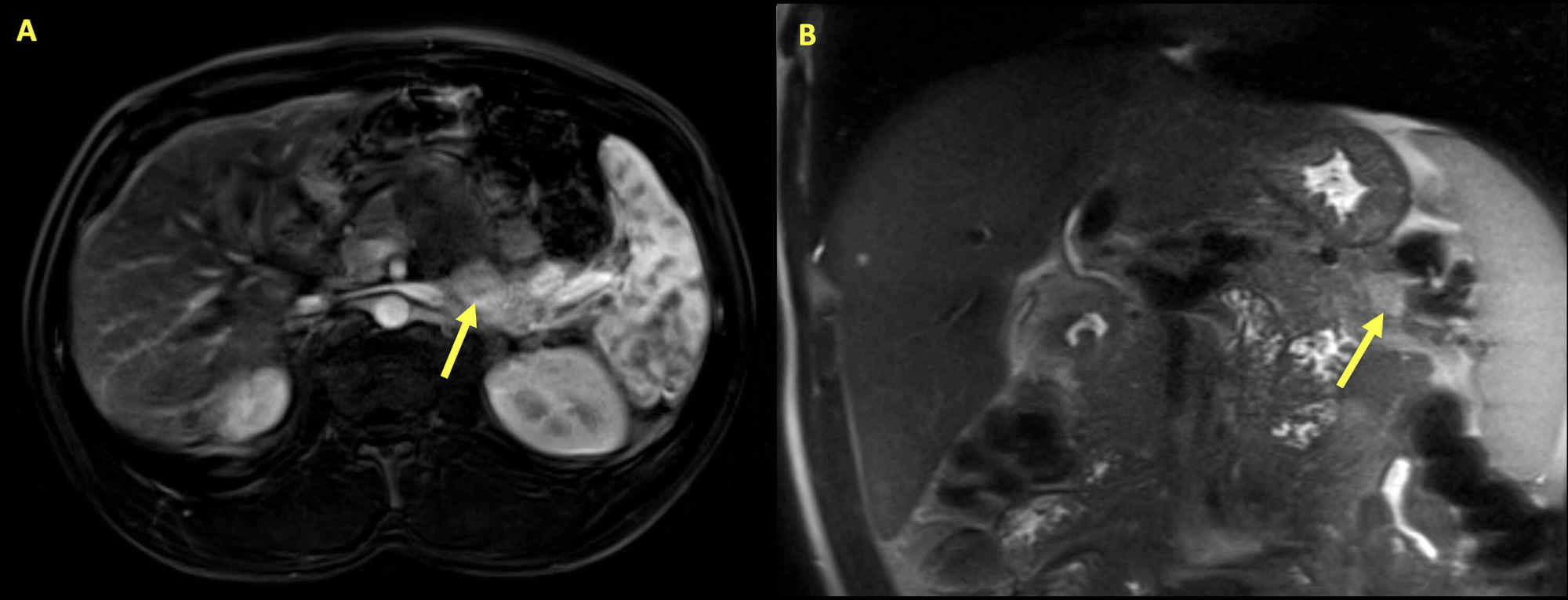
Insulinoma:
Resonancia magnética del abdomen de un hombre de 34 años que presenta hipoglucemia e hipercalcemia. Se observan múltiples lesiones pancreáticas y las flechas resaltan la lesión más grande. La masa es de un área de 2,8 cm x 1,3 cm con realce variable y restricción de la difusión dentro del páncreas, consistente con un insulinoma.
Las pruebas posteriores revelaron una elevación de la hormona paratiroidea intacta. Con el hiperparatiroidismo primario y el insulinoma, se realizaron pruebas genéticas, que mostraron la mutación del gen MEN1 que confirmó la presencia del síndrome NEM1.
Para recordar las localizaciones del desarrollo tumoral en la NEM1, recuerde las 3 P:
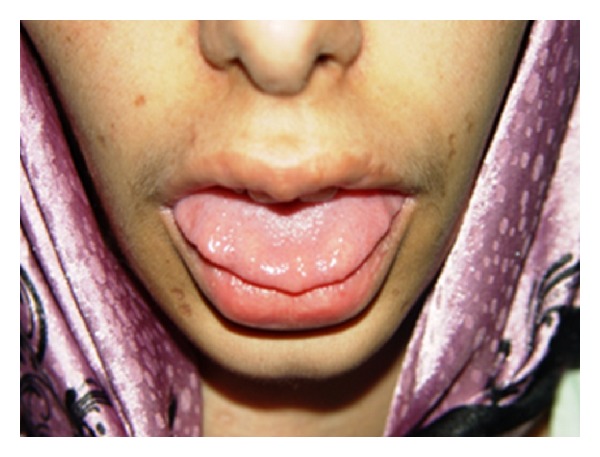
Neoplasia endocrina múltiple (NEM)2B:
La imagen muestra neuromas en el tercio anterior de la lengua en un paciente con NEM2B
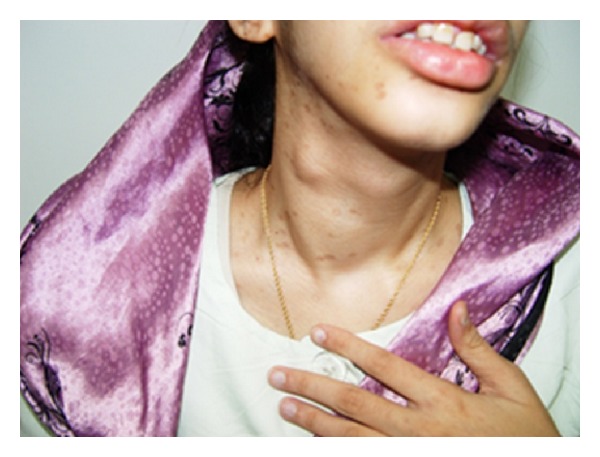
Neoplasia endocrina múltiple (NEM)2B:
La imagen muestra que el paciente tiene un bocio con un nódulo tiroideo en el lóbulo derecho. La biopsia reveló posteriormente un carcinoma medular de tiroides.
Para recordar la localización del desarrollo tumoral en la NEM2, recuerde las 3 P:
