Los LOS Neisseria eritrocitos, o glóbulos rojos, son las células más abundantes de la sangre. Si bien los LOS Neisseria eritrocitos en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el feto se producen inicialmente en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el saco vitelino y luego en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el hígado, la médula ósea es el principal lugar de producción. La eritropoyesis comienza con las células madre hematopoyéticas, que se convierten en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum progenitores comprometidos con el linaje y se diferencian en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum eritrocitos maduros. El proceso se produce por etapas, y la extrusión de los LOS Neisseria núcleos y los LOS Neisseria orgánulos se produce antes de la maduración. Así, los LOS Neisseria eritrocitos maduros carecen de núcleo y tienen una forma bicóncava. Los LOS Neisseria eritrocitos transportan hemoglobina y su forma permite un transporte eficaz del oxígeno. Cada día se producen miles de millones de eritrocitos, ya que su vida media es de aproximadamente 120 días. Los LOS Neisseria eritrocitos que cumplen con su ciclo son eliminados por los LOS Neisseria macrófagos del bazo, el hígado y la médula ósea.
Last updated: Dec 18, 2025
Los LOS Neisseria eritrocitos, también llamados glóbulos rojos, son estructuras terminalmente diferenciadas que carecen de núcleo, pero que están llenas de hemoglobina portadora de oxígeno. Los LOS Neisseria eritrocitos son las células más abundantes de la sangre.
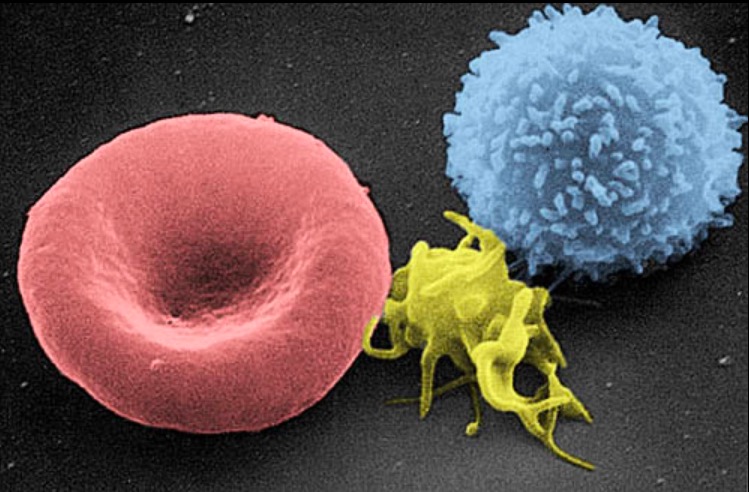
Micrografía electrónica de barrido de una célula sanguínea:
De izquierda a derecha: eritrocito humano, trombocito (plaqueta) y leucocito
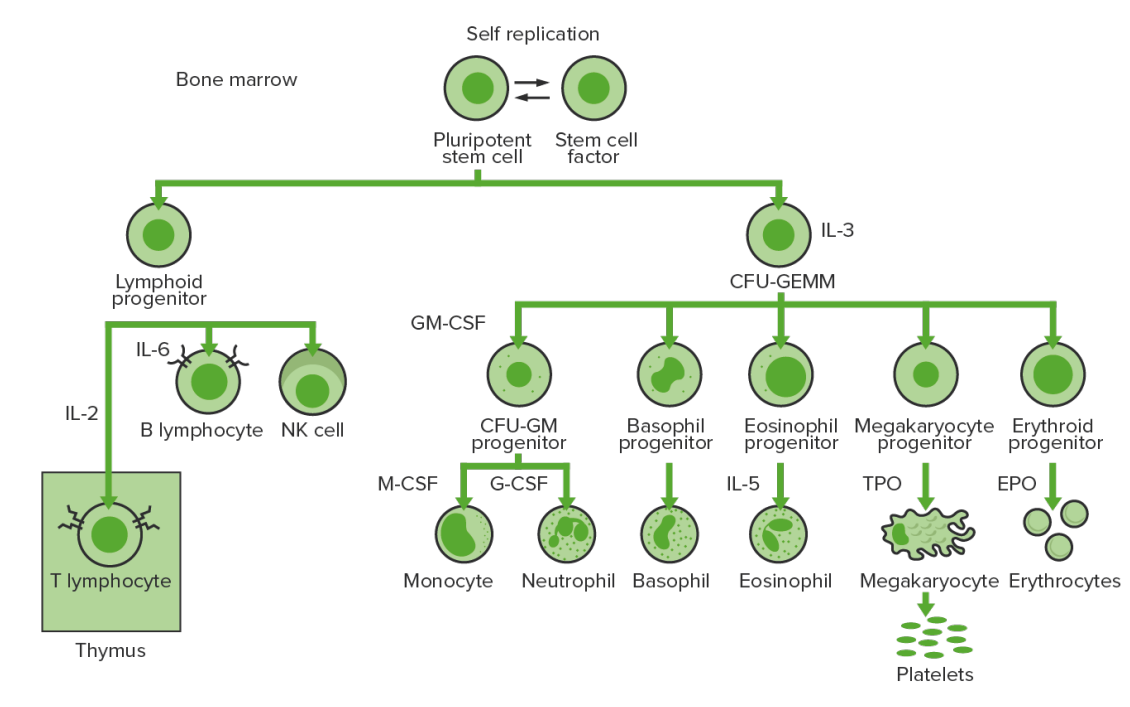
Hematopoyesis de la médula ósea: proliferación y diferenciación de los elementos formados de la sangre.
CFU-GEMM: unidad formadora de colonias de granulocitos, eritrocitos, monocitos y megacariocitos
CFU-GM: unidad formadora de colonias–granulocitos–macrófagos
GM-CSF: factor estimulante de colonias de granulocitos y macrófagos
M-CSF: factor estimulante de colonias de macrófagos
G-CSF: factor estimulante de colonias de granulocitos
NK: Linfocito Natural Killer
TPO: trombopoyetina
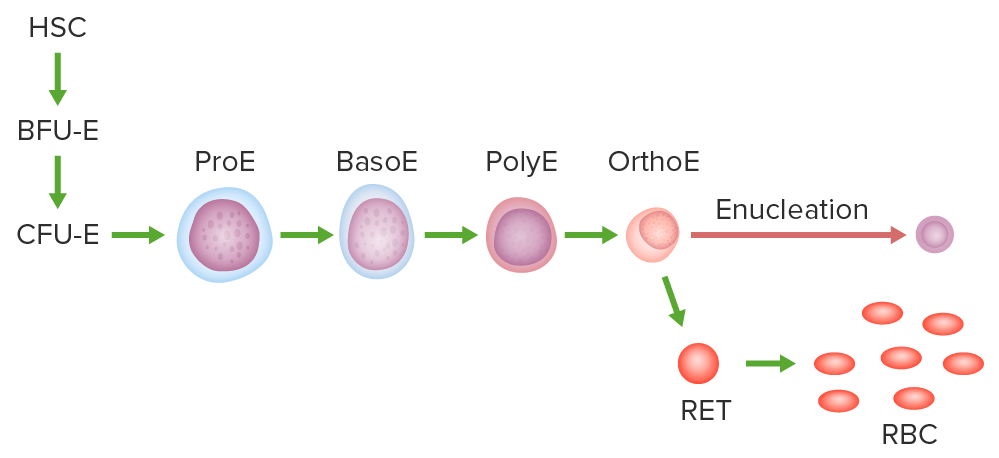
Vía de la eritropoyesis:
Célula madre hematopoyética, unidad formadora de colonias eritroides en ramillete o el progenitor más temprano comprometido, unidad eritroide formadora de colonias, proeritroblasto (ProE), basófilo, policromático y eritroblasto ortocromático.
El eritroblasto ortocromático se enuclea y se convierte en un reticulocito. Tras la expulsión o digestión de los orgánulos, se forma el glóbulo rojo maduro.
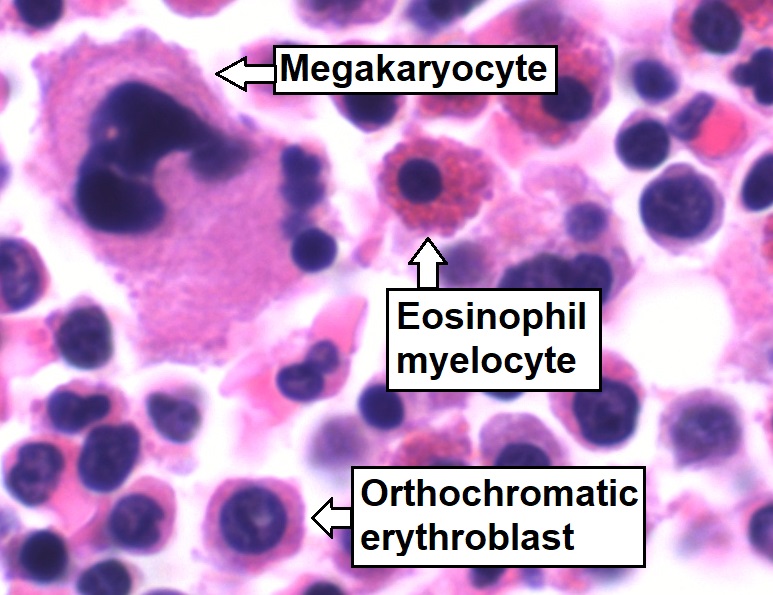
Aspirado de médula ósea que muestra una hematopoyesis trilineal normal:
Células mielomonocíticas (etiquetadas como Eosinophil myelocyte), células eritroides (etiquetadas como Orthochromatic erythroblast) y células megacariocíticas
| Citoquinas y factores de crecimiento | Actividades | Fuente |
|---|---|---|
| Factor de Células Madre (SCF, por sus siglas en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum inglés) | Estimula todas las células progenitoras hematopoyéticas | Células estromales de la médula ósea |
| GM-CSF GM-CSF An acidic glycoprotein of mw 23 kda with internal disulfide bonds. The protein is produced in response to a number of inflammatory mediators by mesenchymal cells present in the hemopoietic environment and at peripheral sites of inflammation. GM-CSF is able to stimulate the production of neutrophilic granulocytes, macrophages, and mixed granulocyte-macrophage colonies from bone marrow cells and can stimulate the formation of eosinophil colonies from fetal liver progenitor cells. GM-CSF can also stimulate some functional activities in mature granulocytes and macrophages. White Myeloid Cells: Histology | Estimula las células progenitoras mieloides | Células endoteliales, células T |
| EPO EPO Glycoprotein hormone, secreted chiefly by the kidney in the adult and the liver in the fetus, that acts on erythroid stem cells of the bone marrow to stimulate proliferation and differentiation. Erythrocytes: Histology | Estimula la eritropoyesis, incluida la diferenciación | Riñón, hígado |
| IL-3 | Mitógeno para todas las células progenitoras de granulocitos y megacariocitos/eritrocitos | Células T colaboradoras |
Otros factores: