La enfermedad de Niemann-Pick, también conocida como deficiencia de esfingomielinasa, es un trastorno de depósito lisosomal hereditario raro. La enfermedad se clasifica sobre la base de la mutación genética. El tipo A y el tipo B resultan de mutaciones en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el gen SMPD-1, lo que resulta en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum una deficiencia de la enzima esfingomielinasa ácida. El tipo C resulta de mutaciones en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure NPC1 o NPC2, que son necesarios para el transporte intracelular de lípidos. Estas mutaciones dan como resultado lisosomas que no pueden metabolizar correctamente la grasa, como la esfingomielina y el colesterol, lo que da como resultado una acumulación progresiva de lípidos intracelulares y daño orgánico. Las manifestaciones clínicas pueden incluir retraso del crecimiento, hepatoesplenomegalia, trombocitopenia, enfermedad pulmonar intersticial, deterioro cognitivo y motor Motor Neurons which send impulses peripherally to activate muscles or secretory cells. Nervous System: Histology, y manchas maculares de color rojo cereza. La neurodegeneración progresiva y una esperanza de vida corta se observan en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la enfermedad de Niemann-Pick tipo A, mientras que enfermedad de Niemann-Pick tipo B generalmente no es neurogénico. El diagnóstico de la enfermedad de Niemann-Pick se basa en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la sospecha clínica y puede confirmarse mediante la medición de la actividad de esfingomielinasa o biomarcadores, pruebas genéticas o biopsia. Actualmente, no existe una cura para la enfermedad de Niemann-Pick, por lo que el tratamiento es de soporte y se enfoca en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum controlar los LOS Neisseria síntomas.
Last updated: Dec 15, 2025
La enfermedad de Niemann-Pick es un trastorno de depósito lisosomal autosómico recesivo que se clasifica según la mutación genética y la deficiencia enzimática:
Lisosomas normales:
En EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la enfermedad de Niemann-Pick tipo A y B:
En EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la enfermedad de Niemann-Pick tipo C:
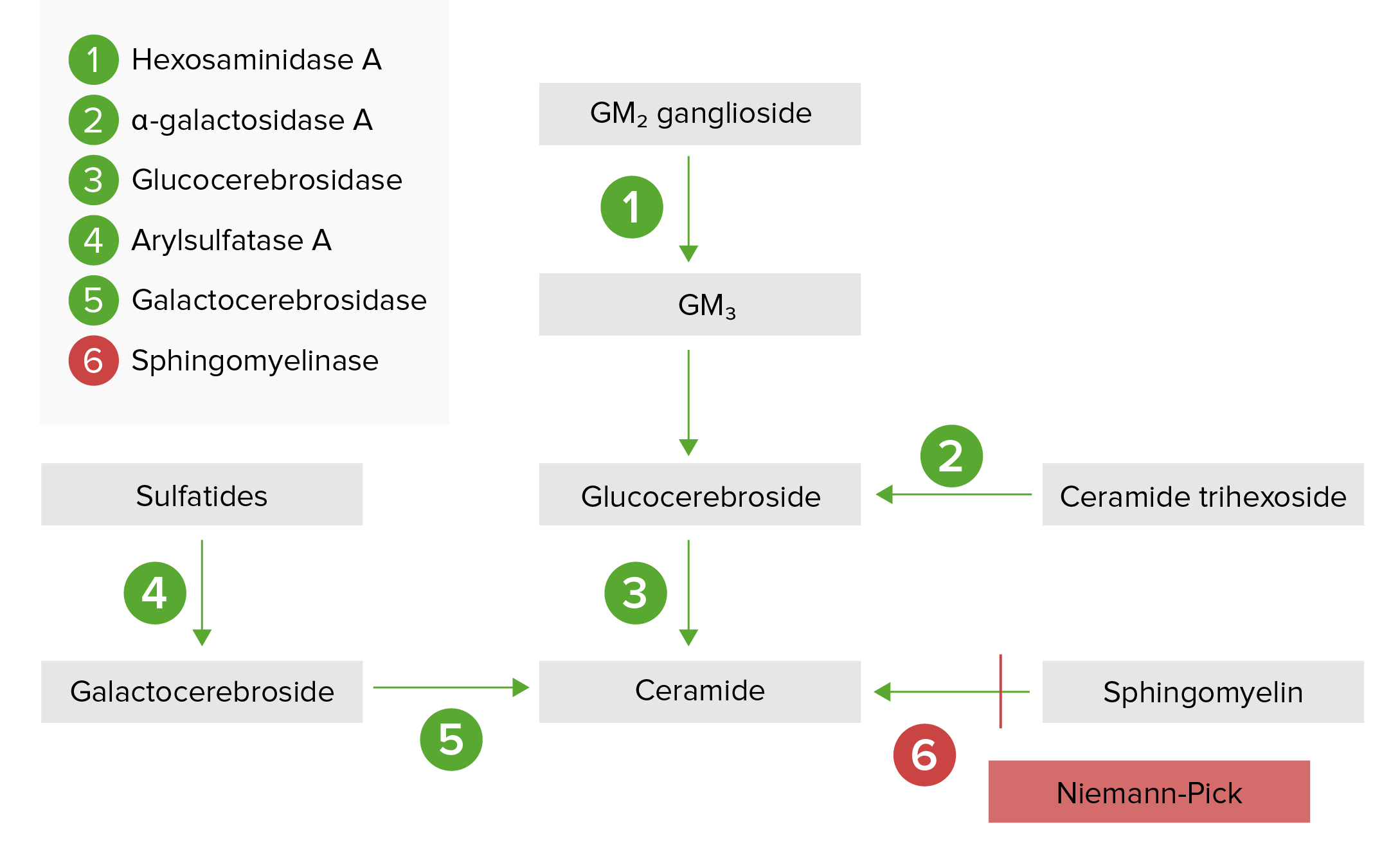
La vía de depósito lisosomal:
La enfermedad de Niemann-Pick (tipos A y B) es el resultado de la deficiencia de esfingomielinasa ácida, lo que resulta en una acumulación de esfingomielina.
La presentación clínica de la enfermedad de Niemann-Pick depende del tipo y la severidad de la enfermedad.
Curso de la enfermedad:
Signos y síntomas:
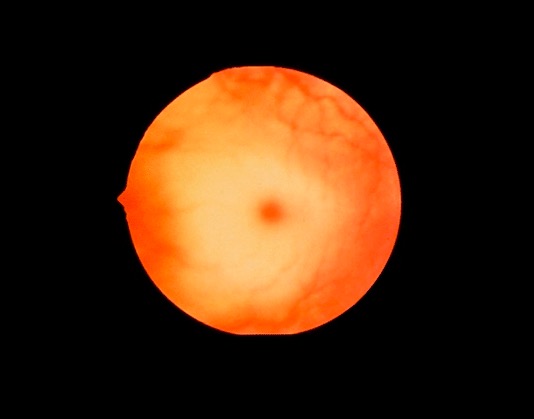
Imagen de fondo de ojo que muestra una mancha rojo cereza, que puede verse en la enfermedad de Niemann-Pick
Imagen: “Cherry red spot” por Jonathan Trobe, MD Licencia: CC BY 3.0Curso de la enfermedad:
Signos y síntomas:
Curso de la enfermedad:
Signos y síntomas:
La sospecha de enfermedad de Niemann-Pick se plantea sobre la base de las características clínicas. El diagnóstico se puede confirmar con las siguientes investigaciones:
No hay cura para la enfermedad de Niemann-Pick. El tratamiento es de soporte y multidisciplinario.