Las distrofias miotónicas son trastornos genéticos debidos a mutaciones genéticas autosómicas dominantes y tienen 2 formas clínicas principales: distrofia miotónica tipo 1 y distrofia miotónica tipo 2. Las distrofias miotónicas son enfermedades heterogéneas que afectan principalmente a los LOS Neisseria músculos, pero, a diferencia de otras distrofias musculares, también tienen efectos multisistémicos. Ambos tipos presentan miotonía, debilidad muscular y mialgias; sin embargo, la distrofia miotónica tipo 1 es grave y conlleva una esperanza de vida reducida, mientras que la tipo 2 es leve con una esperanza de vida normal. El diagnóstico se realiza clínicamente, con pruebas genéticas y mediante electromiografía. El tratamiento es principalmente de soporte.
Last updated: Dec 15, 2025
La distrofia miotónica es una enfermedad muscular hereditaria, autosómica dominante, que provoca incompetencia en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la relajación muscular, lo que da lugar a debilidad y desgaste muscular progresivos.
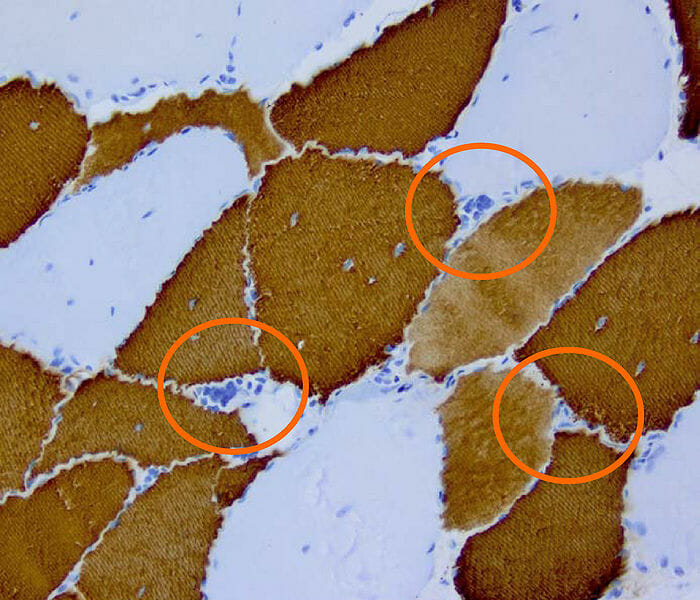
Histopatología de la distrofia miotónica de tipo 2
Imagen: “DM2 Histopathology” por Marvin 101. Licencia: CC BY 3.0Ambos tipos de distrofia miotónica causan miotonía (contracción muscular sostenida) con dificultad o retraso para la relajación después de contraerse.
Síntomas:
Manifestaciones asociadas:
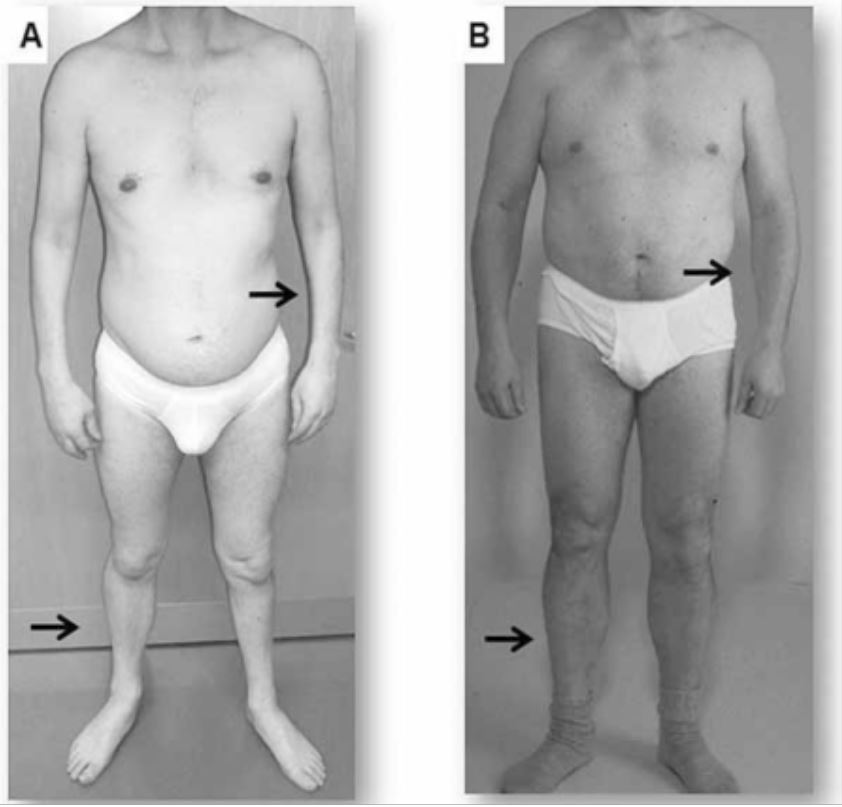
Presentación clínica de dos adultos con distrofia miotónica
A: Atrofia del antebrazo y la pierna en un paciente con distrofia miotónica tipo 1
B: La atrofia no se observa en este paciente con distrofia miotónica tipo 2, en el que los síntomas tienden a ser leves y proximales.