Las enfermedades de almacenamiento lisosómico son un grupo de trastornos metabólicos causados por mutaciones genéticas en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum las enzimas responsables de la función lisosómica normal. La disfunción de los LOS Neisseria procesos enzimáticos provoca una acumulación de metabolitos no digeridos, lo que da como resultado muerte celular. Los LOS Neisseria principales grupos de enfermedades de almacenamiento lisosómico incluyen esfingolipidosis, oligosacaridosis y mucolipidosis. Los LOS Neisseria subgrupos dentro de las principales enfermedades tienen diferentes mecanismos subyacentes y manifestaciones clínicas.
Last updated: Apr 24, 2025
Las enfermedades de almacenamiento lisosómico son condiciones metabólicas raras causadas por mutaciones genéticas de las enzimas lisosómicas, que conducen a un metabolismo disfuncional y a la acumulación de glicosaminoglicanos, glicoproteínas o glicolípidos.
Los LOS Neisseria trastornos se deben a una deficiencia de una hidrolasa lisosómica específica o de las enzimas necesarias para la función lisosómica:
Los LOS Neisseria trastornos son considerados grupos de desórdenes raros del metabolismo intracelular que son heredados individualmente. De los LOS Neisseria 40 trastornos clasificados, 15 representan la mayoría de los LOS Neisseria casos. La categorización es por la acumulación de metabolitos intermediarios:
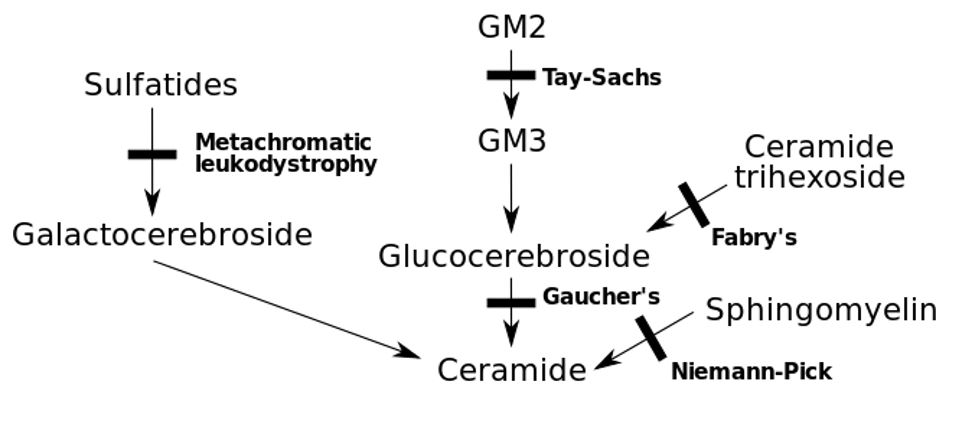
Errores congénitos del metabolismo con el déficit genético asociado
Imagen: “Inborn errors of metabolism” por Huckfinne. Licencia: Dominio Público| Grupo | Subgrupo | Descripción |
|---|---|---|
| Esfingolipidosis | Gangliosidosis GM2 |
|
| Gangliosidosis GM1 | Causada por una mutación en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el gen GLB1 que codifica para la β-galactosidasa-1 | |
| Enfermedad de Gaucher |
|
|
| Enfermedad de Fabry |
|
|
| Leucodistrofia metacromática | Deficiencia en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la actividad de arilsulfatasa A | |
| Enfermedad de Krabbe |
|
|
| Lipogranulomatosis diseminada (enfermedad de Farber) | ||
| Enfermedad de Niemann-Pick |
|
|
| Oligosacaridosis | Sialidosis Sialidosis Overview of Lysosomal Storage Diseases |
|
| Galactosialidosis Galactosialidosis Overview of Lysosomal Storage Diseases |
|
|
| Fucosidosis Fucosidosis An autosomal recessive lysosomal storage disease caused by a deficiency of alpha-l-fucosidase activity resulting in an accumulation of fucose containing sphingolipids; glycoproteins, and mucopolysaccharides (glycosaminoglycans) in lysosomes. The infantile form (type I) features psychomotor deterioration, muscle spasticity, coarse facial features, growth retardation, skeletal abnormalities, visceromegaly, seizures, recurrent infections, and macroglossia, with death occurring in the first decade of life. Juvenile fucosidosis (type II) is the more common variant and features a slowly progressive decline in neurologic function and angiokeratoma corporis diffusum. Type II survival may be through the fourth decade of life. Overview of Lysosomal Storage Diseases |
|
|
| Manosidosis | 2 tipos:
|
|
| Mucolipidosis | Enfermedad de células I |
|
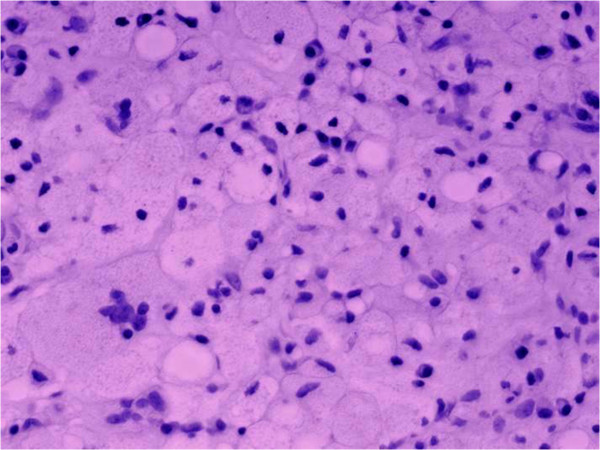
La enfermedad de Gaucher conduce a necrosis ósea:
El tejido conectivo está infiltrado con numerosas células de Gaucher (células reticuloendoteliales cargadas de lípidos y vacuoladas, con citoplasma granular agrandado y núcleos redondos desplazados).
La presentación varía según la etiología del trastorno de almacenamiento lisosómico y puede ocurrir poco después del nacimiento o posteriormente en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la adultez:
| Grupo | Subgrupo | Signos y síntomas |
|---|---|---|
| Esfingolipidosis | Gangliosidosis GM2 |
|
| Gangliosidosis GM1 |
|
|
| Enfermedad de Gaucher |
|
|
| Enfermedad de Fabry |
|
|
| Leucodistrofia metacromática |
|
|
| Enfermedad de Krabbe |
|
|
| Enfermedad de Niemann-Pick | ||
| Oligosacaridosis | Sialidosis Sialidosis Overview of Lysosomal Storage Diseases |
|
| Galactosialidosis Galactosialidosis Overview of Lysosomal Storage Diseases |
|
|
| Fucosidosis Fucosidosis An autosomal recessive lysosomal storage disease caused by a deficiency of alpha-l-fucosidase activity resulting in an accumulation of fucose containing sphingolipids; glycoproteins, and mucopolysaccharides (glycosaminoglycans) in lysosomes. The infantile form (type I) features psychomotor deterioration, muscle spasticity, coarse facial features, growth retardation, skeletal abnormalities, visceromegaly, seizures, recurrent infections, and macroglossia, with death occurring in the first decade of life. Juvenile fucosidosis (type II) is the more common variant and features a slowly progressive decline in neurologic function and angiokeratoma corporis diffusum. Type II survival may be through the fourth decade of life. Overview of Lysosomal Storage Diseases |
|
|
| Mucolipidosis | Enfermedad de células I |
|
El diagnóstico se basa en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la enfermedad específica de almacenamiento lisosómico, pero es importante comprender algunos principios generales:
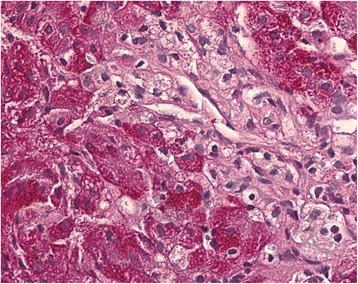
Una célula de Niemann-Pick de una muestra de hígado que muestra células de Kupffer inflamadas con citoplasma espumoso, que es típico de la enfermedad de Niemann-Pick tipo C
Imagen: “Histopathological liver biopsy findings” por Degtyareva AV, Mikhailova SV, Zakharova EY, Tumanova EL, Puchkova AA. Licencia: CC BY 4.0El tratamiento depende del trastorno específico. Los LOS Neisseria principios generales del tratamiento incluyen:
Es posible que se requiera una derivación para personas con hidrocefalia debida a un trastorno de almacenamiento lisosómico
.
Imagen por Lecturio.Mientras que los LOS Neisseria trastornos de almacenamiento lisosómico abarcan un amplio espectro de enfermedades, varias otras condiciones pueden superponerse a los LOS Neisseria trastornos. Algunas condiciones son subgrupos dentro del espectro de los LOS Neisseria trastornos de almacenamiento lisosómico: