Los LOS Neisseria anticonvulsivantes son agentes farmacológicos utilizados para lograr el control de las convulsiones y/o prevenir episodios convulsivos. Los LOS Neisseria anticonvulsivantes abarcan varios medicamentos con diferentes mecanismos de acción, incluido el bloqueo de los LOS Neisseria canales iónicos (Na+ y Ca CA Condylomata acuminata are a clinical manifestation of genital HPV infection. Condylomata acuminata are described as raised, pearly, flesh-colored, papular, cauliflower-like lesions seen in the anogenital region that may cause itching, pain, or bleeding. Condylomata Acuminata (Genital Warts)2+) y la inhibición de la recaptación de ácido gamma aminobutírico ( GABA GABA The most common inhibitory neurotransmitter in the central nervous system. Receptors and Neurotransmitters of the CNS, por sus siglas en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum inglés). El fenobarbital, la fenitoína, la carbamazepina, el ácido valproico y la etosuximida son los LOS Neisseria anticonvulsivos de 1ra generación. Los LOS Neisseria medicamentos anticonvulsivantes generalmente tienen una farmacocinética complicada, múltiples interacciones farmacológicas y rangos terapéuticos estrechos en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum comparación con los LOS Neisseria medicamentos de nueva generación.
Last updated: Dec 15, 2025
Los LOS Neisseria medicamentos anticonvulsivantes se usan para suprimir la actividad eléctrica anormal en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el cerebro a través de varios mecanismos.
El estado de hiperexcitabilidad de las neuronas se produce a través de los LOS Neisseria siguientes 3 pasos:
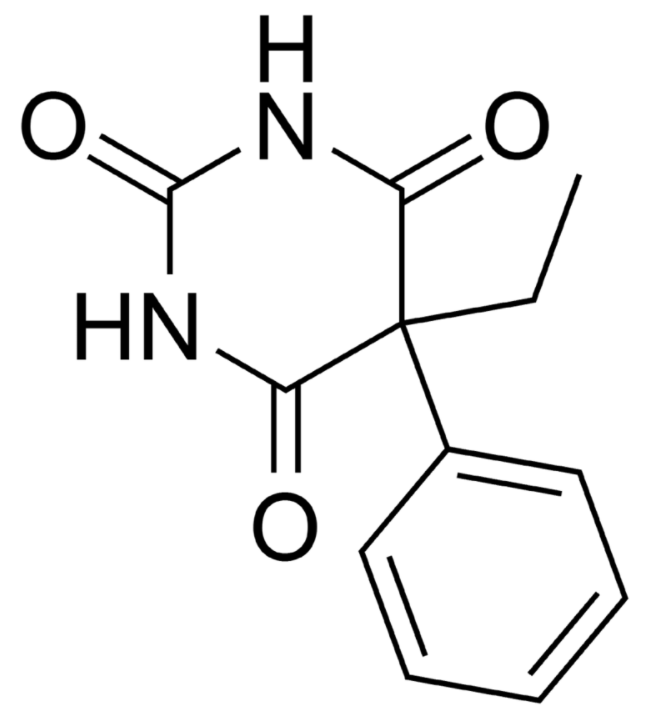
Estructura química del fenobarbital
Imagen : “Phenobarbital” por Harbinary. Licencia: dominio público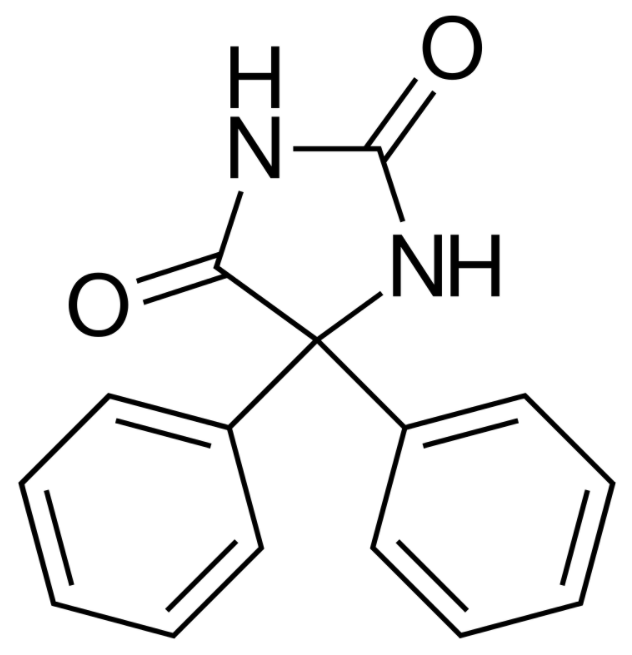
Estructura química de la fenitoína
Imagen : “Phenytoin structure” por Harbin. Licencia: Dominio Público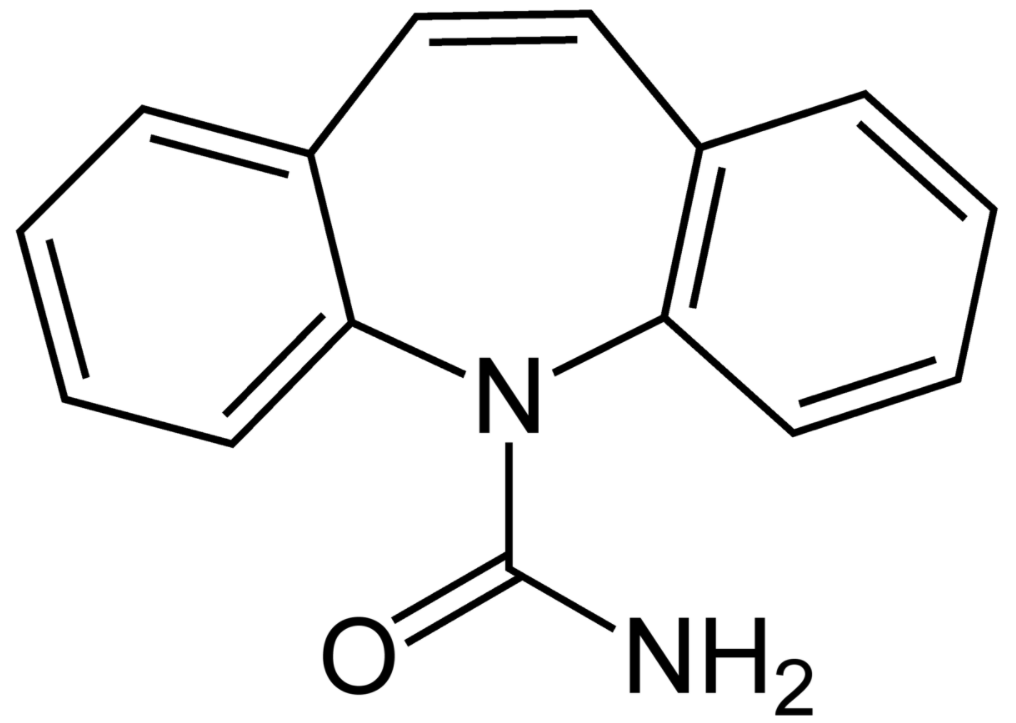
Estructura química de la carbamazepina
Imagen : “Carbamazepine structural formulae” por Jü. Licencia: Dominio Público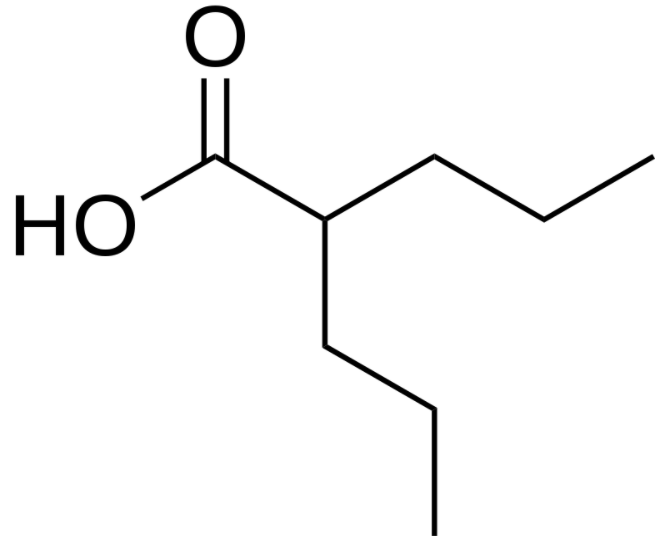
Estructura química del ácido valproico
Imagen: “Valproic acid” por Harbin. Licencia: Dominio Público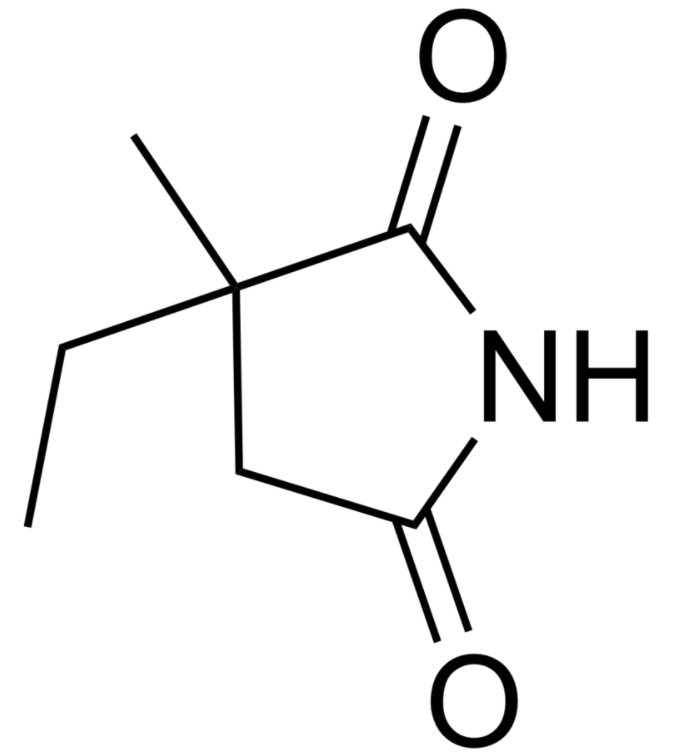
Estructura química de la etosuximida
Imagen: “Ethosuximide” por Fvasconcellos. Licencia: Dominio PúblicoTratamiento de las crisis de ausencia
| Medicamentos | Mecanismo de acción | Principales efectos secundarios | Interacciones | Indicaciones |
|---|---|---|---|---|
| Barbitúricos (fenobarbital) | Se une a las subunidades del receptor Receptor Receptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell. Receptors GABA GABA The most common inhibitory neurotransmitter in the central nervous system. Receptors and Neurotransmitters of the CNS → ↑ actividad inhibidora de GABA GABA The most common inhibitory neurotransmitter in the central nervous system. Receptors and Neurotransmitters of the CNS |
|
Induce CYP y UGT | Convulsiones generalizadas y focales |
| Fenitoína | Bloquea los LOS Neisseria canales de Na+ dependientes de voltaje |
|
Induce CYP y UGT |
|
| Carbamazepina | Bloquea los LOS Neisseria canales de Na + |
|
Induce CYP y UGT |
|
| Ácido valproico |
|
|
Inhibe CYP y UGT |
|
| Etosuximida | ↓ Corrientes de canal Ca CA Condylomata acuminata are a clinical manifestation of genital HPV infection. Condylomata acuminata are described as raised, pearly, flesh-colored, papular, cauliflower-like lesions seen in the anogenital region that may cause itching, pain, or bleeding. Condylomata Acuminata (Genital Warts) 2+ tipo T | Somnolencia (depresión del SNC) | ↑ Efectos de la fenitoína | Crisis de ausencia |