Playlist
Show Playlist
Hide Playlist
Phagocytic Cell Disorders – Primary Immunodeficiency

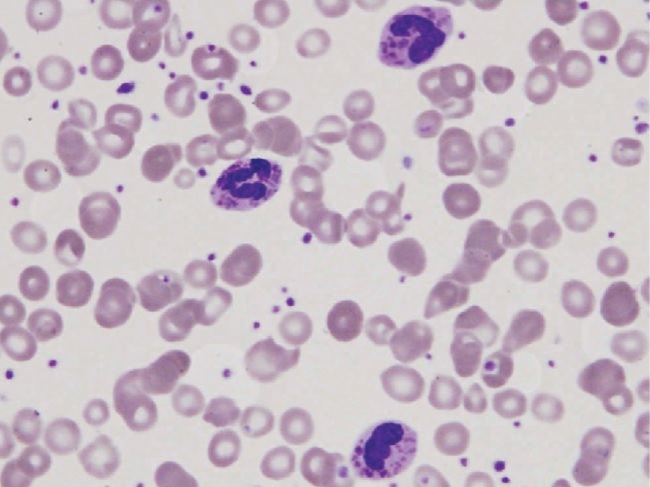
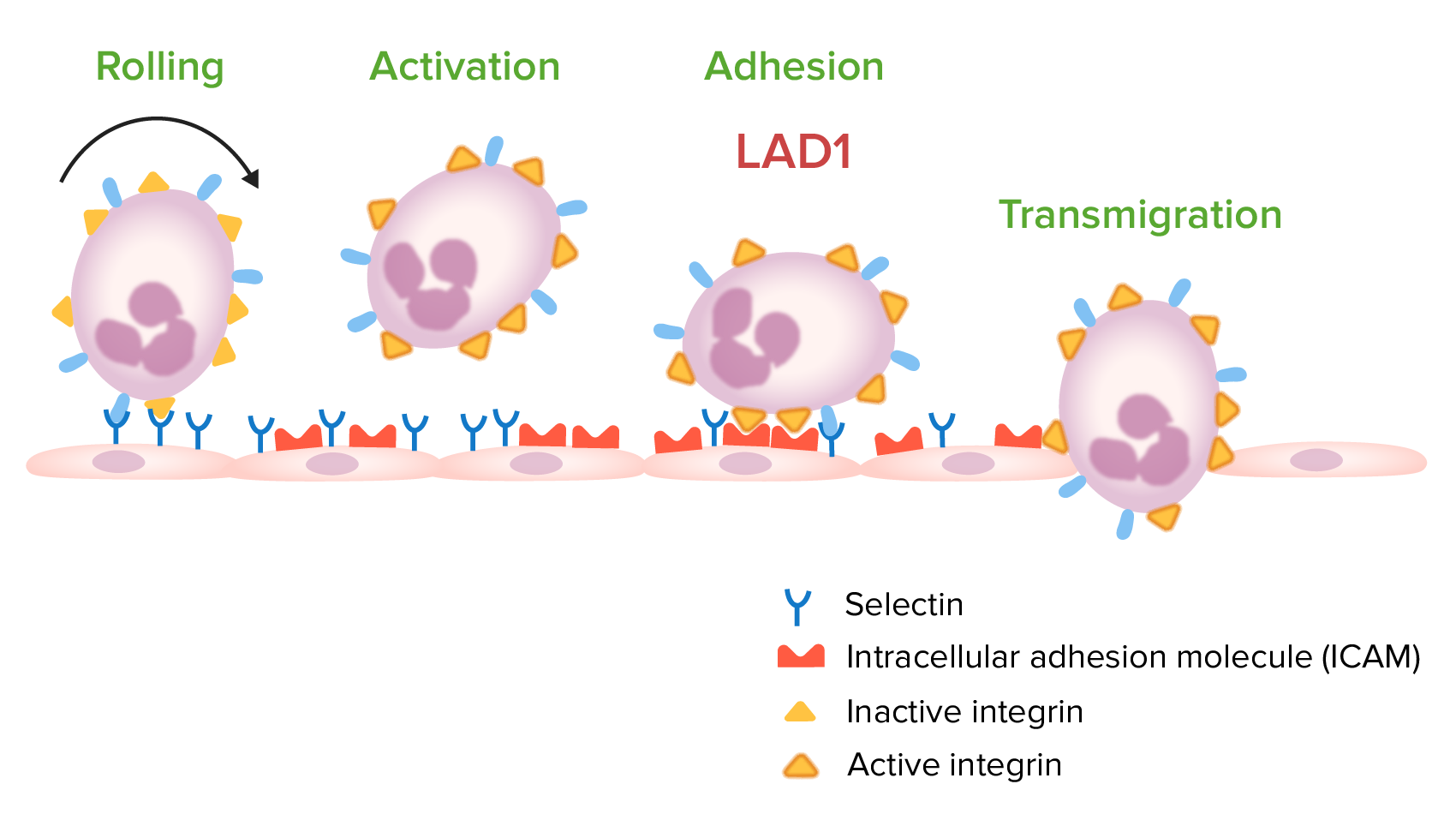
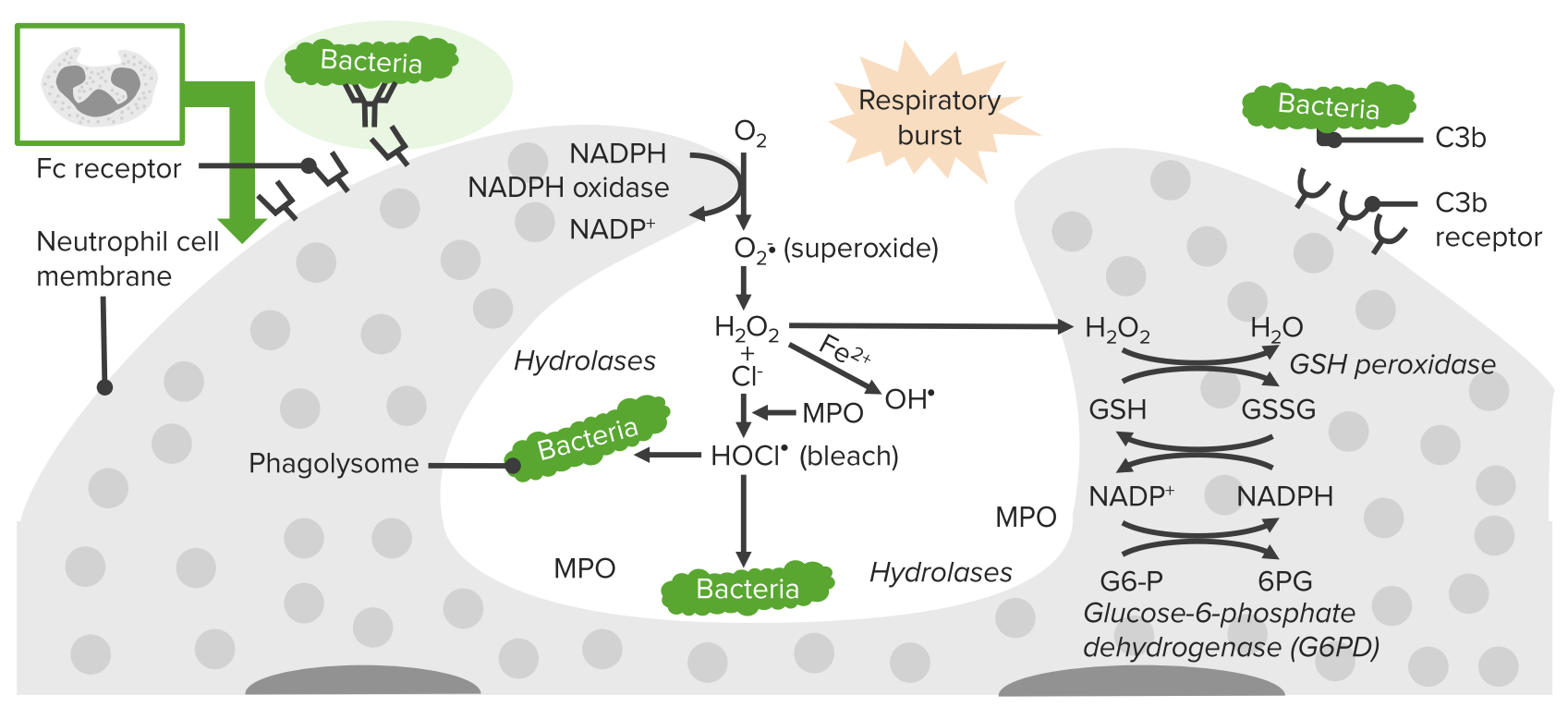
-
Slides Immunodeficiency.pdf
-
Reference List Immune System.pdf
-
Download Lecture Overview
00:01 There are a number of gene defects that can affect phagocytic cells, and these are listed here. 00:08 Defective genes for components of the NADPH oxidase can result in chronic granulomatous disease. 00:18 And here we see a number of typical infections, with an increased instance and severity of disease with Staph aureus, Aspergillus fumigatus, Candida albicans and so on. 00:31 A defect in the CD18 β-subunit; CD18 is an integrin adhesion molecule, leads to leukocyte adhesion deficiency type I. We see an increased instance of pyogenic bacteria. In contrast, a defective gene for the GDP-fucose transporter leads to leukocyte adhesion deficiency or LAD II; again, with an increased incidence of pyogenic bacteria. Kindlin 3 deficiency leads to leukocyte adhesion deficiency type III, again with an increased incidence of pyogenic bacteria. 01:11 And then a defect in the LYST gene leads to the disorder called Chediak-Higashi syndrome. 01:17 And a range of Staphylococci and Streptococci, and other species are seen with an increased incidence in this condition. 01:27 Let’s have a look in a little bit more detail at chronic granulomatous disease. 01:32 In the vast majority of patients, this is due to a defect in subunits of the nicotinamide adenine dinucleotide phosphate oxidase, the NADPH oxidase. 01:42 It affects monocytes, macrophages and neutrophils. 01:48 And they fail to produce reactive oxygen intermediates that are required to kill engulfed microorganisms. 01:56 The NADPH oxidase consists of a number of different subunits as you can see here. 02:04 The function of this oxidase is to produce reactive oxygen species that are involved in killing engulfed microorganisms. 02:13 A defect in the gene encoding the gp91-phox component of the NADPH oxidase, is the X-linked form of this disease because that gene is present on the X chromosome. 02:29 The genes for the other components of the NADPH oxidase are found on the non-sex chromosomes, in other words, the autosomes. 02:37 And the autosomal recessive p22-phox, p47-phox, p40-phox or p67-phox variants of chronic granulomatous disease are caused by gene defects in these particular autosomal genes. 02:58 In a minority of patients, rather than having gene defects in components of the NADPH oxidase, there can be genetic mutations in the myeloperoxidase or glucose-6-phosphate dehydrogenase genes. 03:14 This leads to a similar but less severe phenotype in these patients. 03:21 Turning now to leukocyte adhesion deficiency; as we’ve already heard, there are three types. 03:29 LAD type I is due to a lack of the CD18 β subunit of the β2 integrins. 03:36 LAD type II is due to defective GDP-fucose transporter, and therefore an inability to fucosylate sialyl Lewis structures. 03:50 Whereas LAD type III is due to a mutation in the integrin activation molecule, kindlin 3. 03:58 So any one of these three gene defects, in different ways can lead to defective adhesion of leukocytes and compromise the ability to fight infection. 04:12 In Chediak-Higashi syndrome, there’s a defect in the LYST (lysosomal trafficking) gene. 04:19 There’s an accumulation of giant intracytoplasmic granules. 04:23 This is due to defective migration of the late endosomal/lysosomal compartment within the cell, which interferes with the correct function of these cells. 04:34 There is dysfunction of neutrophils, of natural killer cells, and of cytotoxic T-cells. 04:41 And here we can see a neutrophil with these giant granules accumulating within the cell, and this compromises the function of the cell. 04:50 Likewise, in the natural killer cell and in the cytotoxic T-cell, these large granules accumulate in the cytoplasm and interfere with the correct activity of the cell. 05:02 Patients suffer from a range of pyogenic infections, particularly with Staph aureus, Strep pyogenes, Pneumococci, Aspergillus species and Pseudomonas aeruginosa.
About the Lecture
The lecture Phagocytic Cell Disorders – Primary Immunodeficiency by Peter Delves, PhD is from the course Immunodeficiency and Immune Deficiency Diseases. It contains the following chapters:
- Defects of Phagocytic Cells
- Chronic Granulomatous Disease
- Leukocyte Adhesion Deficiency
- Chediak-Higashi Syndrome
Included Quiz Questions
X-linked chronic granulomatous disease (XL-CGD) arises due to mutations in which of the following genes?
- gp91phox
- p22phox
- q26-qter
- Xq28
- 19q13.1
Which of the following conditions is caused by mutations in the lysosomal trafficking regulator (LYST) gene?
- Chediak-Higashi syndrome
- Leukocyte adhesion deficiency II
- Chronic granulomatous disease
- Glucose-6-phosphate dehydrogenase deficiency
- Leukocyte adhesion deficiency III
Which of the following defects causes leukocyte adhesion deficiency type 3?
- Kindlin 3
- Cluster of differentiation 18 β-subunit
- Guanosine 5′-diphospho-β-L-fucose transporter
- Fucosylation of sialyl Lewis X structure
- Nicotinamide adenine dinucleotide phosphate oxidase
Chediak-Higashi syndrome most strongly affects which types of immune cells?
- Neutrophils and macrophages
- B lymphocytes and plasma cells
- Helper T cells
- Regulatory T cells
- Dendritic cells
Author of lecture Phagocytic Cell Disorders – Primary Immunodeficiency

Peter Delves, PhD
Customer reviews
5,0 of 5 stars
| 5 Stars |
|
5 |
| 4 Stars |
|
0 |
| 3 Stars |
|
0 |
| 2 Stars |
|
0 |
| 1 Star |
|
0 |
