Playlist
Show Playlist
Hide Playlist
Disorders of Amino Acid Metabolism

-
Slides MetabolicDisorders Pediatrics.pdf
-
Download Lecture Overview
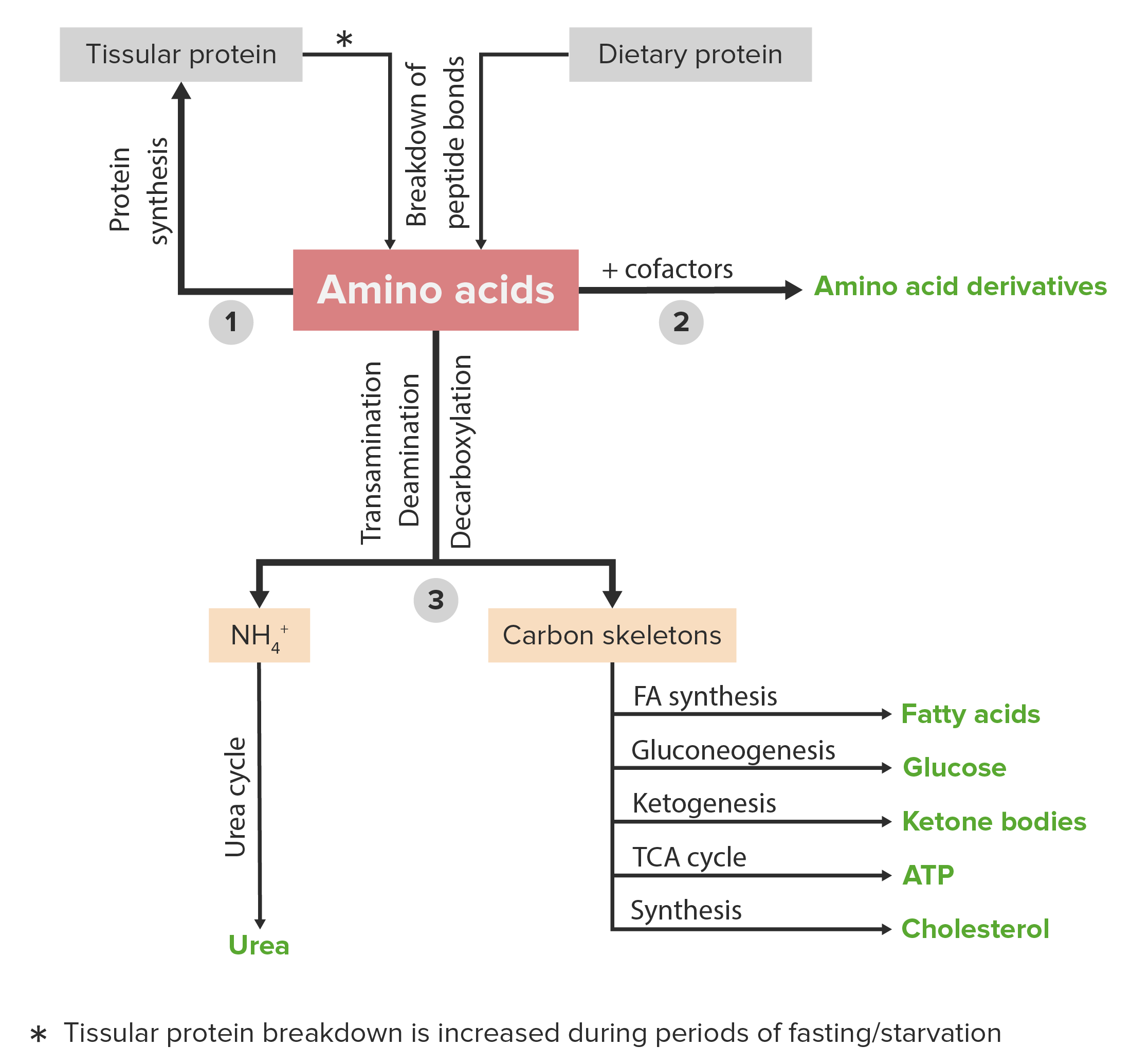
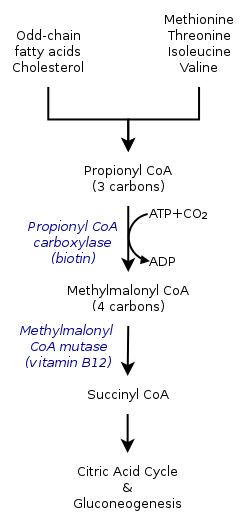
00:01 Let's switch gears from sugar to proteins and specifically focus on amino acid metabolism. 00:09 Now we recall some of the amino acids, typically the branch chain ones are called organic acids and we'll refer to these two diseases slightly different. 00:20 The amino acid metabolism defects and the organic acidemias. 00:24 In the amino acid metabolisms, these are typically auto resembles autosomal recessive as are the organic acidemias. 00:32 But the amino acids that are involved are different, so typical amino acid metabolism defects include problems with metabolizing phenylalanine and tyrosine. 00:44 And the organic acidemias are those branch chain amino acids like leucine, isoleucine and valine. 00:51 Just say leucine, isoleucine, and valine a few times over and over again and you'll start to remember those are the organic acidemias. 00:57 Let's start with the amino acids. 01:01 The most common inborn error of metabolism regarding amino acids is PKU or phenylketonuria. 01:08 PKU is something we screen for in every state in the United States on the newborn screen because avoidance of the amino acid phenylalanine, which is the problem here, is incredibly important to improving their outcome. 01:24 These children have a difficulty in metabolizing phenylalanine. 01:28 This is not that uncommon, 1 in 15,000 US children. 01:33 So we pick this up on the newborn screen because we can intervene with a low phenylalanine diet. 01:40 A special formula which is lacking of phenylalanine or has a very tiny, tiny amount so that they do get some necessary for building their proteins but never too much because we don't want them having to break these down. 01:54 They can't. 01:56 These children, if undetected, and sometimes even if detected will develop developmental delay. 02:02 They can have infantile spasms, which are very severe seizures which are hard to treat. 02:07 They can have all kinds of behavior problems, they may have microcephaly, they may have seizures, and what may show up on your exam is a mousy urine smell which is how this was previously detected before we got the newborn screen. 02:24 There are other disorders of amino acid metabolism that are very different in the way they present. 02:30 One example is homocystinuria or homocysteinemia. 02:35 These patients have a hard time handling that amino acid. 02:39 Homocysteine in the blood can result in increased risk of blood clots and hypercoagulability. 02:46 Interestingly, these patients can also have slipped lenses as you can see in this picture. 02:51 Another defect is Alkaptonuria. 02:55 What's key for your exam is brownish urine. 02:58 These patients will have a brownish urine that is one of the ways we might pick it up. 03:03 Although this is usually done in the newborn screen as well. 03:06 Another example would be hereditary tyrosinemia. 03:09 So these are all examples of specifically amino acid metabolism defects. 03:14 Let's switch now to the organic acidemias. 03:19 And the most common problem which is an organic acidemia in the United States is probably maple-syrup urine disease that has such a unique name that is probably easier to remember. 03:31 Maple-syrup urine disease is called this because it has a very sweet smell, almost like maple syrup to the urine. 03:38 And what's interesting about maple-syrup urine disease is it's much more common in the American Amish population. 03:44 The Amish live in these areas of this map here. 03:48 The Amish have an instance of approximately 1 in 400 of children getting maple-syrup urine disease. 03:55 Again, there's that sweet scent to the urine and this is one of the branch chain amino acids. 04:01 So this is an organic acid problem. 04:03 This acid, because of the enzyme defect, can't enter the Krebs cycle so patients do worse during periods of catabolism. 04:12 Catabolism, remember, is when you need your protein to make some sugar. 04:17 So they're going to break down their proteins so that they can get over the pyruvate and build sugar during a period of fasting. 04:24 During these periods of fasting, the patients are breaking down their proteins into individual amino acids and because they can't metabolize this one, they start to develop an increase in their levels in the blood which can then result in problems. 04:44 These problems include hypoglycemia, because they're not able to make sugar very well, which results also in seizure. 04:51 They may develop vomiting and they generally have a neurologic decline. 04:55 This is damaging their brain so we need to keep these patients in a state where they are not undergoing catabolism. 05:02 Typically, when we admit these children to the hospital during say, gastroenteritis, we will give them continuous glucose as a way to sort of control that catabolic state. 05:13 So there are many types of organic acidemias. 05:18 Maple-syrup urine disease is one of them. 05:21 We also have isovaleric acidemia, proprionic acidemia, and MMA or methylmalonic acidemia. 05:30 These are all types of organic acidemias. 05:34 Now, they are all a little bit different and this can be a bit dizzying to remember. 05:40 So I'm putting up these slides so you can pause the video and try and memorize some of the key aspects of these four branch chain amino acid problems. 05:50 The key points I put it in bold because I think these are most likely to show up on your test. 05:56 Specifically, they like to talk about the maple-syrup urine disease having that maple syrup odor. 06:03 Remember these patients can get cerebral edema, and we typically treat them with thiamine which is different than the others which are usually treated with carnitine. 06:12 Isovaleric acidemia has that sweaty foot odor, they develop hepatomegaly and were gonna treat them with carnitine and glycine. 06:21 Proprionic acidemia, they get alopecia, they desquamation of their skin, and they can develop corneal ulcers in the eye. 06:29 They also develop hepatomegaly and we treat them with carnitine. 06:32 And MMA, they can get very significant hepatosplenomegaly. 06:37 They can get acidosis and high levels of ammonia. 06:40 They can develop cardiomyopathy and renal damage. 06:43 And we treat them with vitamin B12 which is an important cofactor as well as carnitine. 06:48 So that's my summary of these organic acidemias.
About the Lecture
The lecture Disorders of Amino Acid Metabolism by Brian Alverson, MD is from the course Pediatric Endocrinology. It contains the following chapters:
- Disorders of Amino Acid Metabolism
- Amino Acid Metabolism Defects
- Organic Acidemia
Included Quiz Questions
Which of the following amino acid disorders can present with lens dislocation?
- Homocystinemia
- Phenylketonuria
- Alkaptonuria
- Cystinuria
- Hereditary Tyrosinemia
A young infant presents with developmental delay, severe seizures, microcephaly, and a mousy urine smell. What is the most likely diagnosis?
- Phenylketonuria
- Maple Syrup Urine disease
- Homocysteinuria
- Isovaleric acidemia
- Methylmalonic acidemia
A patient with maple syrup urine disease is admitted to the hospital for community acquired pneumonia. Apart from treating his pneumonia with antibiotics, which of the following additional therapy is required for this patient?
- Continuous glucose infusion
- Supplemental potassium
- Half strength normal saline
- Albumin infusion
- Blood transfusion
Which of the following is the most appropriate treatment option for methylmalonic acidemia?
- Carnitine and vitamin B12
- Carnitine only
- Carnitine and glycine
- Vitamine B12 only
- Thiamine
Author of lecture Disorders of Amino Acid Metabolism

Brian Alverson, MD
Customer reviews
5,0 of 5 stars
| 5 Stars |
|
1 |
| 4 Stars |
|
0 |
| 3 Stars |
|
0 |
| 2 Stars |
|
0 |
| 1 Star |
|
0 |
Excellent lecture. It's a complex topic and once more it is made understandable and memorable. Thanks!
