Playlist
Show Playlist
Hide Playlist
Pathophysiology and Clinical Manifestations – Calcific Aortic Stenosis

-
Slides Valvular Hypertensive Heart Disease.pdf
-
Reference List Pathology.pdf
-
Download Lecture Overview
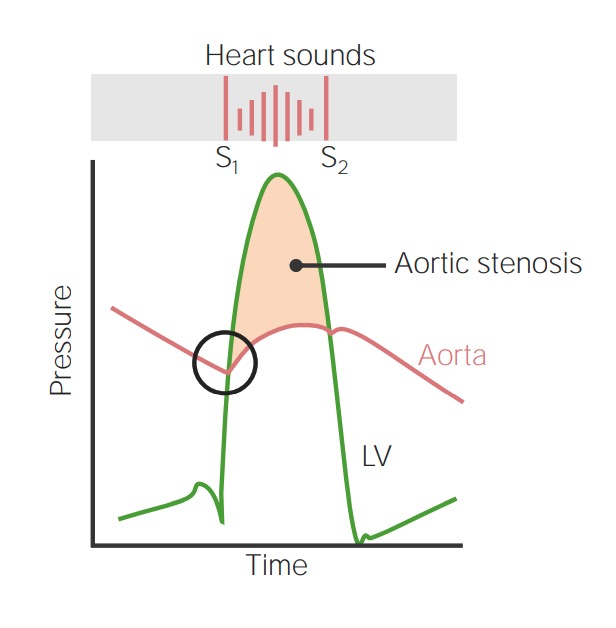
00:01 So, let's talk about calcific valvular degeneration. 00:05 So, this is showing you a normal set of valves in the heart, but we're going to focus here in a minute on mainly the aortic valve where most calcific degeneration happens. 00:16 If you stop and think about it, valves are subjected to really high levels of repetitive mechanical stress. 00:24 You, sitting comfortably at home, your heart is beating 60 beats per minute. 00:29 Me, after three cups of coffee this morning, my heart is beating at 90, but it's got to do that all the time, and those valves are flapping every 60 to 90 times a minute. 00:40 If you count them up, that's 30 to 40 million contractions per year. That's pretty remarkable. 00:48 And during that process, the valve has to get completely out of the way to allow flow to occur, and that's going to snap back and hold the pressure from going retrograde. 00:59 So, there's substantial tissue deformation during each contraction. 01:03 I will say that we had never synthesized synthetic material that will hold up for that many contractions even for just one year. 01:12 All the synthetic material that we've devised fails mechanically within about six months. 01:19 So, about 20 to 30 million contractions. There's also a high transvalvular pressure gradient. 01:27 So, we go from 120 millimeters of mercury at peak systole down to about 80, but you have to have the valve be able to, see, tolerate 120 to open, 80 to close, and snap close and hold. So, that's actually a lot of work, and we're asking those valves to do quite a bit. So, they're under a lot of stress. 01:50 It's remarkable that they have evolved. 01:52 Tissue been - tissue engineered and do so well for the lifetime of most of us. 01:59 So, what does cause this senile calcific degeneration of the aortic stenosis? What's the pathogenesis of that? The factors are felt to be somewhat similar to those that cause atherosclerosis. 02:11 So, high lipids, high blood pressure, inflammation, cigarette smoke, and other things are felt to be somehow contributory. 02:22 But as we'll talk about in a minute, if we try to limit things with, say, hyperlipidemia by giving the patient statin, that does not impact the propensity to calcify. 02:34 So, we don't know exactly, we just know that these risk factors are in some way related. 02:39 When the valve degenerates, the cells that are within the valve, the valvular interstitial cells, die, and on their surface, they start accumulating calcium. 02:52 And so, you get this kind of rock development on the kind of crystallized node of a dead valvular interstitial cell. 03:02 As the cells die and begin to calcify because of the calcium in the system, we also have phosphate come in, and we get deposition of a hydroxyapatite. 03:12 Basically, it's bone-like material and that calcification is rock hard. 03:18 Indeed, the valvular interstitial cells that when they die, may become the kind of crystalline nidus for developing calcification. 03:30 The ones that don't die sense all that bone-like material around them, and they can undergo a phenotypic change, and they can product bone matrix proteins that will, in turn, lead to faster deposition of calcium salts and further degeneration. 03:47 So, those valvular interstitial cells are important players. When they die, they calcify. 03:53 And the ones that live and survive, they see that calcification, they go, I think I should be turning into bone. And they become little bone-like structures. 04:04 So, it turns out, as I already mentioned, interventions that improve the atherosclerotic risk, that is the, say, statins that reduce cholesterol levels, don't appear to significantly impact valvular calcific generation. 04:17 We do not currently have good drugs to prevent this process. 04:23 This is what it looks like. So, we are sitting up in the aorta looking down on an aortic valve. 04:29 Normally, there should be three cusps, and normally, they should be kind of clean looking cuspal tissues. 04:35 And you see those nodules, that's all calcification. 04:39 Those are rocks of absolutely solid rock calcium hydroxyapatite formation. 04:46 This valve will open, but it's going to be begrudgingly opened because of all of these firm rock formations. 04:55 So, this is going to have a functional stenosis. 04:58 If we open up the valve and look at it, you can see the calcifications and they accumulate on the outflow surface of the valve and limit the excursion of the valve. 05:08 The lunula, the free edge, tends not to be involved. 05:12 So, calcific aortic stenosis is the most common of all valvular abnormalities. 05:17 A tricuspid valve is present, as I said many slides ago, in 99% of the population. 05:24 So, in a tricuspid valve, the stenosis usually occurs in the 7th to 9th decades of life. 05:31 On the other hand, if you are so unfortunate as to have a bicuspid aortic valve, which is 1% of us walking around, your stenosis, your calcific degeneration, will occur one to two decades earlier. How do we get bicuspid aortic valve? Well, it is a developmental issue, and we have some genetic understanding. 05:51 So, there are associations that include loss of function, that's what LOF is, loss of function mutations in a NOTCH1 gene. This is a signaling molecule. 06:02 But that's actually a very small percentage of people with bicuspid aortic valve and we don't entirely understand why this happens at such a high frequency overall in the population. 06:15 What happens to result of aortic stenosis? So, the normal aortic valve, when it's open, has a total surface area of about 4 cm2. 06:27 That's pretty substantial of a normally opened aortic valve. 06:31 With calcific degeneration, that - you see the valves become kind of thickened and stiff, and now, instead of having 4 centimeters, you have 0.5 to 1 cm2. 06:43 That's with significant disease. That's where you become symptomatic, is in that range. 06:48 But that really has narrowed the orifice by a lot and you can see then why blood flow through that is going to be impeded. 06:57 As a result of that, now, we have a much higher-pressure gradient. 07:00 The left heart has to squeeze much more aggressively to provide blood flow out the aorta through that tight orifice. 07:09 So, instead of the normal gradient, we have a gradient across the valve of about 75 to 100 millimeters of mercury. 07:18 Normally, there would be a gradient of 20 to 40. 07:21 So, that is substantially more, 3 to 5 times as much. 07:26 And the left ventricular pressure, instead of being 120 millimeters max, can get as high as 200 millimeters of mercury or more. 07:36 So, there's increased pressure that the heart is generating. 07:40 In order to accomplish that, you have to have myocyte hypertrophy. 07:44 The heart, the individual myocytes will increase the number of sarcomeres and they lay them down side by side by side, so that's in parallel, and that makes for a concentric hypertrophy that allows for greater force generation. 08:00 But at the same time, we are now increasing the capillary density. 08:05 The microvasculature is not substantially changing. 08:08 So, you get these big old cardiac myocytes with the same capillary density trying to profuse them, and in the middle of that big old cardiac myocyte, there's going to be relative areas of ischemia due to increased diffusion distance. 08:22 So, you're going to be more prone to having ischemia and arrythmias as a result of the hypertrophy. 08:28 Secondly, the increased pressures that are being generated are also going to have an effect on atherosclerosis development. 08:36 As a result of this ischemic - relative ischemia of the cardiac myocytes and atherosclerosis, you can have cardiac decompensation. 08:47 And there are three symptoms that herald a very bad prognosis. 08:52 So, if a patient with aortic stenosis develops angina or congestive heart failure or has syncopal episodes, passes out, loses consciousness, that actually portends death within two to five years. 09:07 And in fact, it's more like two years. So, this is a bad disease. 09:11 Aortic stenosis kills faster than many of the worse cancers that you know about. 09:17 So, this is a bad actor. What do we do for this? Well, really, the only therapy we have now, because we don't know how to prevent the calcification just yet, is a surgical valve replacement.
About the Lecture
The lecture Pathophysiology and Clinical Manifestations – Calcific Aortic Stenosis by Richard Mitchell, MD, PhD is from the course Valvular and Hypertensive Heart Disease.
Included Quiz Questions
Which of the following risk factors is NOT related to calcific valvular degeneration?
- Elevated serum calcium levels
- Hyperlipidemia
- Hypertension
- Inflammation
- Smoking
Which aspect of the aortic valve system is typically affected by calcific degeneration?
- Outflow surface of the aortic valve
- The inner (ventricular) surface of the aortic valve
- Lunula (free edge) of the aortic valve
- Wall of the proximal aorta
- All surfaces of the aortic valve
Which of the following statements about patients with bicuspid aortic valves is TRUE?
- Stenosis due to calcifications occurs 1–2 decades earlier than in those with normal aortic valves.
- It occurs in 5%–10% of the population.
- Stenosis typically occurs during the seventh to ninth decades of life.
- It is not associated with the NOTCH1 genetic mutation.
- It is more commonly found in women.
At what aortic valve orifice diameter do most patients become symptomatic from calcific aortic stenosis?
- 0.5–1 cm²
- 4 cm²
- 0.1 cm²
- 8 cm²
- 2 cm²
What three conditions or symptoms are associated with increased mortality within 2–5 years in patients with calcific aortic stenosis?
- Syncope, angina, congestive heart failure
- Angina, syncope, pulmonary hypertension
- Pulmonary hypertension, diabetes, syncope
- Angina, endocarditis, arrhythmia
- Arrhythmia, syncope, congestive heart failure
What is the current treatment for symptomatic calcific aortic valve stenosis?
- Surgical valve replacement
- Diuretics
- ACE-inhibitors/ARBs
- Heart transplant
- Calcium channel blockers
Author of lecture Pathophysiology and Clinical Manifestations – Calcific Aortic Stenosis

Richard Mitchell, MD, PhD
Customer reviews
5,0 of 5 stars
| 5 Stars |
|
3 |
| 4 Stars |
|
0 |
| 3 Stars |
|
0 |
| 2 Stars |
|
0 |
| 1 Star |
|
0 |
3 customer reviews without text
3 user review without text
