Playlist
Show Playlist
Hide Playlist
Other Airways Diseases: Summary

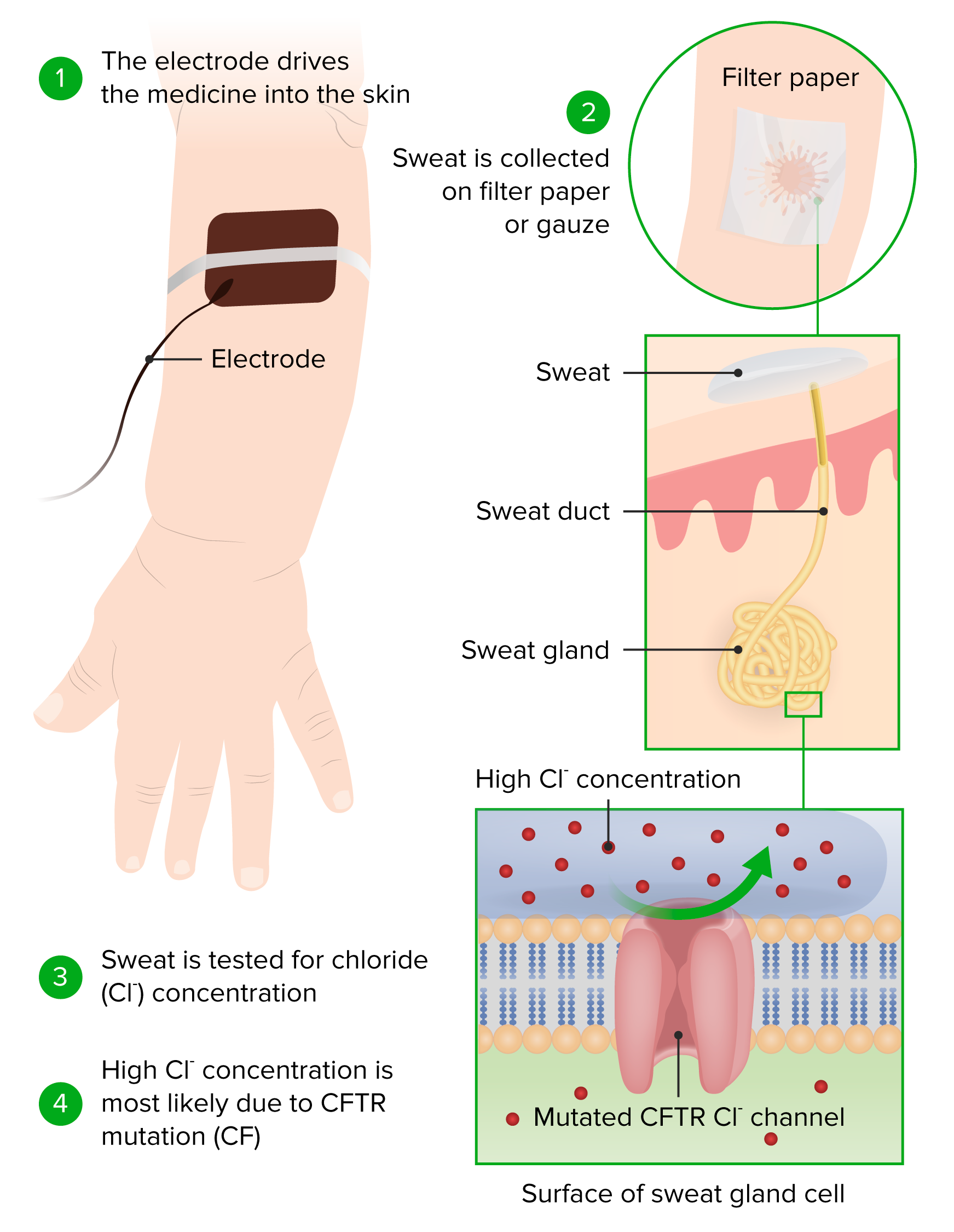
-
Slides 05 MajorAirwaysObstructionBronchiectasis RespiratoryAdvanced.pdf
-
Download Lecture Overview
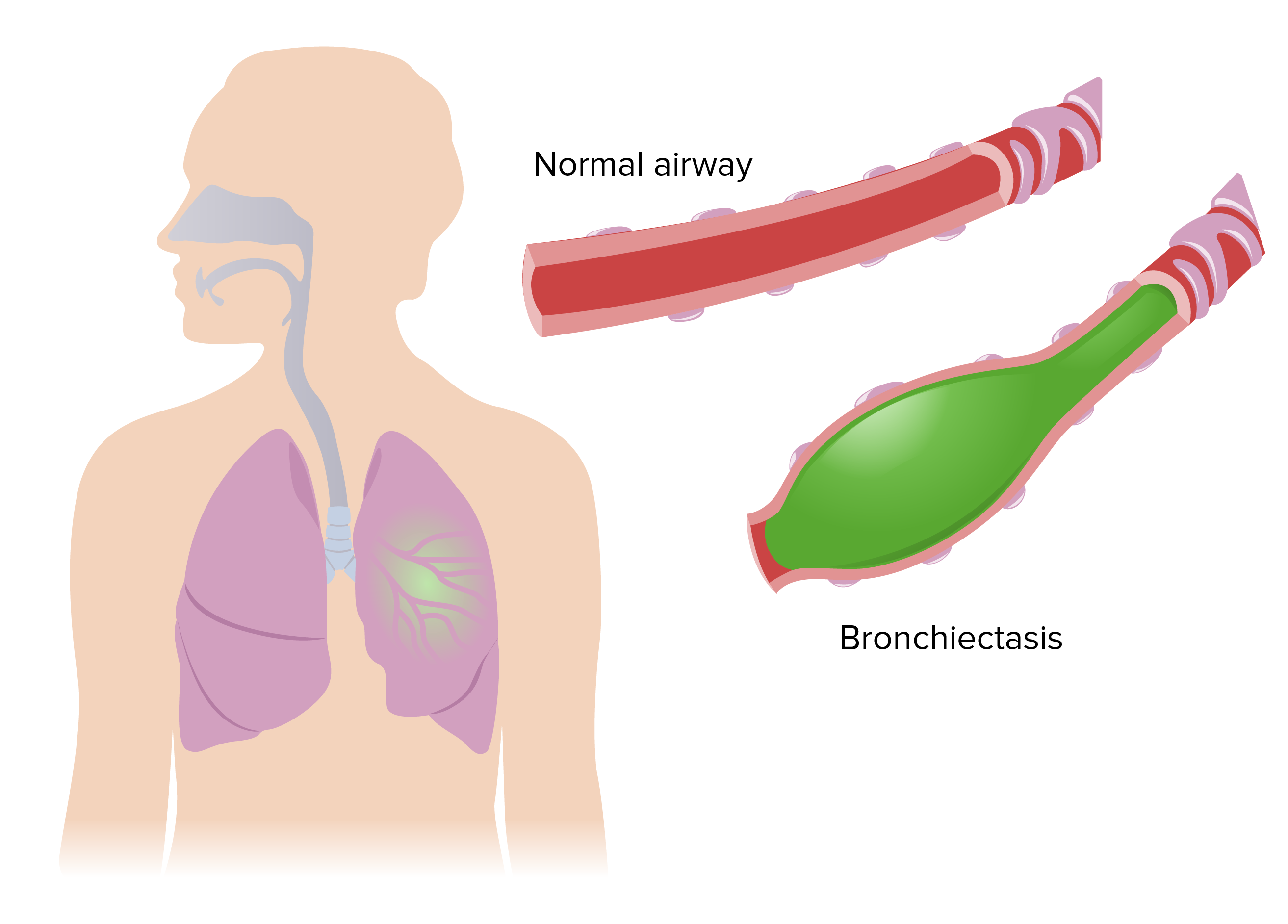
00:00 in patients presenting with major hemoptysis. So, to summarize the discussion of airways disease in this third lecture. Upper airways obstruction is uncommon but can be life threatening and it’s often misdiagnosed as asthma or COPD. Flow volume loops. The CT scan and bronchoscopy can identify patients with upper airways obstructions, but you need to think about doing those tests, you need to consider those diagnosis. Bronchiectasis is common and has multiple different causes, and the main problem is that the patients get recurrent infections and eventually if they get uncontrolled recurrent infections that will lead to progressive airways obstruction and potentially respiratory failure and death. So controlling infections is the main aim in these patients to prevent lung function loss over time. Cystic fibrosis is a specific cause of bronchiectasis an inherited the defect of the CFTR gene, and that causes a very severe bronchiectasis which almost invariably will progress to severe airways obstruction but is associated with multiple other problems such as malabsorbtion, diabetes, infertility and obstruction. Lastly, we've discussed minor hemoptysis which is, the main problem there is whether the patient is maybe having lung cancer or not. And major hemoptysis which is a significant life threatening emergency that requires aggressive treatment. Thank you for listening.
About the Lecture
The lecture Other Airways Diseases: Summary by Jeremy Brown, PhD, MRCP(UK), MBBS is from the course Airway Diseases.
Included Quiz Questions
A patient with a history of tuberculosis presents with hemoptysis of more than 250 ml over the past 24 hours. Which of the following steps is INCORRECT regarding the management of this patient?
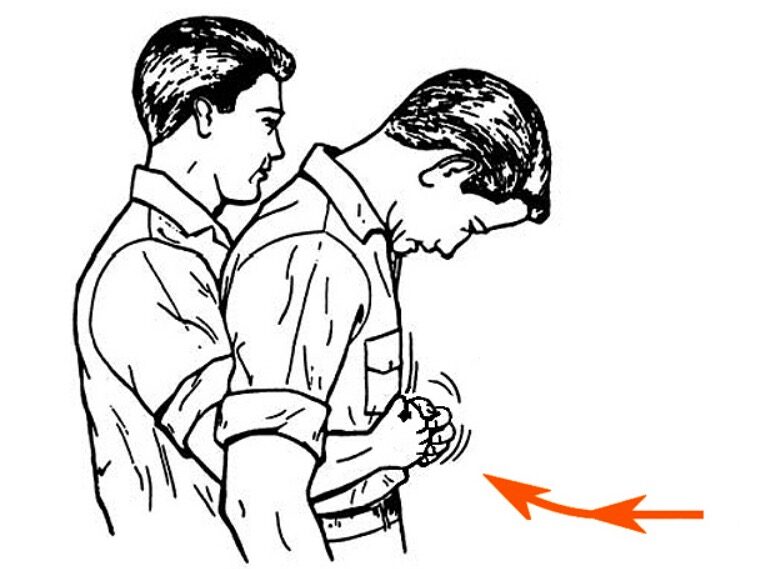
- The patient should lie down on the side of the unaffected lung.
- Bronchial arteriogram and embolization may be indicated.
- Monitoring for hemoglobin levels is recommended.
- Oxygen therapy is usually required.
Which of the following is the most appropriate management step for a patient not responding to initial measures to control massive hemoptysis?
- Arteriography
- Tranexamic acid
- Vitamin K supplementation
- Nasal packing
- Thoracotomy
Author of lecture Other Airways Diseases: Summary

Jeremy Brown, PhD, MRCP(UK), MBBS
Customer reviews
5,0 of 5 stars
| 5 Stars |
|
5 |
| 4 Stars |
|
0 |
| 3 Stars |
|
0 |
| 2 Stars |
|
0 |
| 1 Star |
|
0 |
