Playlist
Show Playlist
Hide Playlist
Cystic Fibrosis (CF): Pathology


-
Slides Cystic Fibrosis.pdf
-
Download Lecture Overview
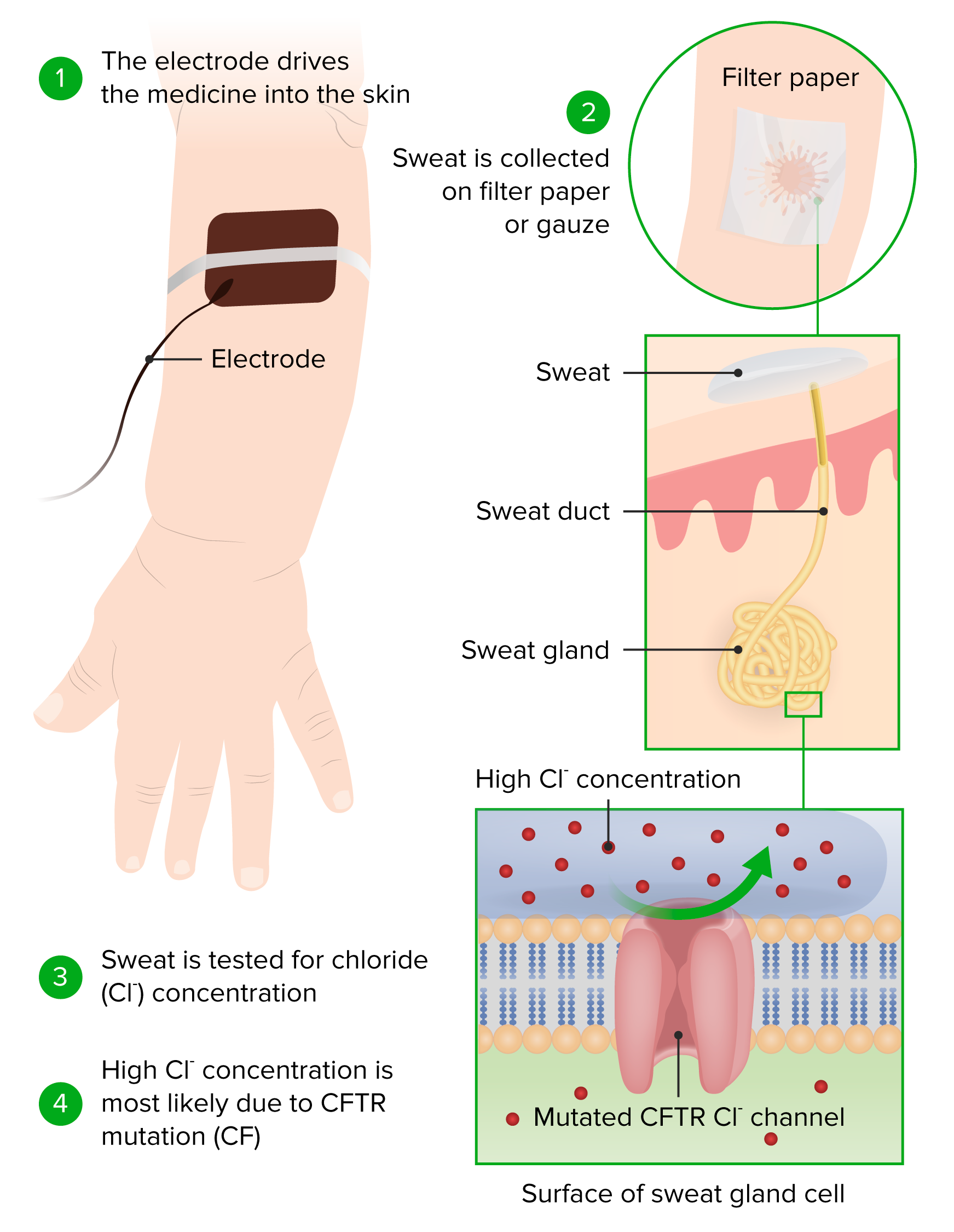
00:01 In this lecture, we are going to talk about cystic fibrosis. 00:06 So when we think of cystic fibrosis, it may be important to step a little bit back and look at some of the epidemiologic facts about the disease. 00:15 We need to remember that 4% of Caucasian people in the United States carry this gene. 00:20 It’s incredibly highly prevalent. 00:24 It’s less common, but absolutely occurs in all ethnic groups. 00:29 I have myself seen cases where a child was of a different ethnicity and there was a delay in the diagnosis because there was a presumption that because this child wasn’t Caucasian that he might not have the illness. 00:42 The disease is autosomal recessive, so while the gene is in 4%, far fewer actually have the illness. 00:51 Fifty percent of the cases are diagnosed before six months of age so many of these children are diagnosed in infancy, some are diagnosed a little bit later on in early childhood, but most is diagnosed by the age of five, virtually all of it, about three-quarters before the age of two. 01:12 So let’s look a little bit at the genetics of the disease and then specifically at the CFTR gene. 01:19 The primary issue in cystic fibrosis is a genetic defect in the cystic fibrosis transmembrane conductance regulator protein or CFTR. 01:30 This protein is found in all exocrine tissues and many epithelial tissues including lung, liver, the GI tract, and the skin. 01:42 So a mutation in this gene, and there are many different mutations that can occur, results in abnormal secretion of chloride in these tissues. 01:53 This can affect the lungs of a patient, the pancreas, the liver, the intestine, and the reproductive tract. 02:03 We’re going to talk about all of these. 02:05 So let’s focus first on pulmonary disease in patients with cystic fibrosis. 02:10 So the idea here is that because these patients have very thick airway secretions as a result of a defect in that chloride transport, these patients are at increased risk for infection. 02:24 They’re more prone to unusual organisms like Pseudomonas aeruginosa, whereas, regular people, who get a bacterial pneumonia, are more likely to have something like Strep pneumo. 02:36 These recurrent infections of unusual organisms result in chronic inflammation of the lung. 02:43 That in turn will destroy the parenchyma of the lung and will result in bronchiectasis. 02:50 This gradual degradation of pulmonary mechanics and pulmonary function is the hallmark of this disease. 02:59 Airways frequently will be colonized. 03:02 Certainly, Staph aureus and increasingly MRSA is colonizing in these airways or things like Haemophilus influenzae. 03:10 But again, because of the thickness of the secretions, these patients can have unusual organisms like Pseudomonas aeruginosa or Burkholderia cepacia. 03:20 Cepacia in particular has a high degree of resistance and may be a negative predictor of mortality. 03:28 These patients with Burkholderia cepacia may be in big trouble. 03:33 Those bugs will grow as a biofilm along the surface of the airway and can be very difficult if not impossible to eradicate. 03:44 The damage from these infections results in lots of different things, such as spontaneous, recurrent pneumothorax, pulmonary hemorrhage, respiratory distress, and eventually death. 03:59 Pancreatic disease in these patients is also very troubling. 04:04 Remember, these duct secretions are very thick in the pancreas, as well. 04:10 This results to an acute or a chronic pancreatitis. 04:14 The pancreatitis results in an inadequate release of lipase and other important enzymes that are responsible for degradation and digestion of food. 04:26 So these patients can have fat malabsorption, they can have poor absorption of fat soluble vitamins A, D, E, and K. 04:35 As a result of their difficulty with absorbing fat, they often have poor weight gain. 04:42 They can also develop an endocrine disorder in their pancreas because of the damage and can get CF-related diabetes, which is a lot like type 1 diabetes. 04:56 The intestinal disease in these children is very bad as well. 05:01 These patients develop thickened intestinal secretions. 05:05 This rather disgusting substance you see on the slide is meconium and we sometimes see a delay in the passage in meconium in newborns that looks like that, and a delay in the passage of that meconium is highly concerning for an underlying diagnosis of cystic fibrosis. 05:24 Older children can have a complete or partial small bowel obstruction from the thickened stools. 05:32 Infants can get a meconium ileus, which is where the meconium doesn’t pass at all and there’s actually an obstruction. 05:39 In patients with chronic constipation, they can develop rectal prolapse, where the rectal vault actually extrudes out past the rectum and there’s an inversion of the colon outside the body that has to be pushed back in again. 05:55 Patients with very severe distal obstruction can develop DIOS or distal intestinal obstruction syndrome, where they actually are completely incapable of passing stools because of this distal obstruction. 06:09 And remember, any infant who hasn’t passed a stool within 48 hours is at increased risk of having cystic fibrosis. 06:18 Patients with cystic fibrosis may also develop liver disease. 06:24 In patients like this, and you can see here in this picture some fibroblasts and some hepatic degradation, these patients may develop a focal biliary cirrhosis, which causes an underlying liver failure. 06:40 Thick biliary secretions slow down the flow of bile and cause a backup, especially around the biliary tree. 06:50 This also can result in gallstones and gallstone formation and patients may require a cholecystectomy. 06:59 They may get bile duct dilatation as a result of these gallstones, which in turn can increase liver enzymes through backflow and congestion resulting in hepatomegaly and increased liver enzymes. 07:15 Ten percent of cystic fibrosis patients die ultimately of liver disease rather than pulmonary disease. 07:26 There are many other problems that can occur in patients with cystic fibrosis, and during the course of this lecture, I unfortunately can only touch upon a few of them. 07:37 In males, infertility is almost ubiquitous. 07:43 Males usually have a problem with an absence or an inadequacy of the vas deferens, which results in inability to conduct the sperm effectively outside of the male penis. 07:58 In females, they may also have infertility, but usually have normal fertility and pregnancy outcomes are actually usually excellent and they are excellent with appropriate nutrition. 08:13 Remember, what’s key in these patients is the GI disease and the malabsorption as a result of pancreatic insufficiency. 08:21 It is incredibly important for a pregnant woman to gain appropriate weight so that the fetus inside can grow and develop effectively. 08:30 Patients with cystic fibrosis who are pregnant need extra special care to track weight gain and make sure that’s going well. 08:40 Patients with cystic fibrosis are at increased risk for fractures because of poor bone mineralization. 08:47 Remember that vitamin D is one of the fat soluble vitamins; D, E, A, K, and vitamin D at low levels will cause poor bone mineralization and put them at risk for fractures. 09:01 Another issue can be renal stones. 09:04 Renal stones happen because patients with fat malabsorption will hold on to fat inside the lumen of the gut. 09:13 That fat will now saponify with calcium. 09:19 That calcium is usually binding oxalate and now oxalate can be absorbed and extruded in the renal system, and these patients may get oxalate stones. Fat saponifies calcium leading oxalate to be reabsorbed in increased amounts. The nephrolithiasis in cystic fibrosis is mostly due to hyperoxaluria. Patients with cystic fibrosis also can get venous thrombosis in the kidney as well.
About the Lecture
The lecture Cystic Fibrosis (CF): Pathology by Brian Alverson, MD is from the course Pediatric Pulmonology. It contains the following chapters:
- Epidemiology
- CFTR
- Pulmonary Disease
- Pancreatic Disease
- Intestinal Disease
- Liver Disease
- Other Problems
Included Quiz Questions
Which bacterium is a serious problem in children with cystic fibrosis?
- Pseudomonas aeruginosa
- Group A streptococcus
- Pneumocystis jiroveci
- B. cereus
A patient with cystic fibrosis most likely inherits the disease from...?
- Both mother and father.
- Father.
- Mother.
- Paternal grandparents.
- Maternal grandparents.
A 15-year-old boy with cystic fibrosis suffers from a recurrent pulmonary infection. Which of the following is the end stage complication of his condition?
- Bronchiectasis
- Tuberculosis
- Bronchogenic carcinoma
- Asbestosis
- Sarcoidosis
Which of the following liver pathologies is least likely to occur in patients with Cystic Fibrosis?
- Hepatocellular carcinoma
- Focal biliary cirrhosis
- Gallstones
- Thick biliary secretions
- Hepatomegaly
Author of lecture Cystic Fibrosis (CF): Pathology

Brian Alverson, MD
Customer reviews
4,5 of 5 stars
| 5 Stars |
|
1 |
| 4 Stars |
|
1 |
| 3 Stars |
|
0 |
| 2 Stars |
|
0 |
| 1 Star |
|
0 |
Very clear and interesting. Also very well explained.I think the video is very well lectured.
One of the typical "pediatric" disease. Also hits multiple systems. It's good that there are several lectures dedicated to it. Thank you!
