Playlist
Show Playlist
Hide Playlist
Resulting Condition: Tay-Sachs Disease

-
Slides Cellular Pathology - Degradation.pdf
-
Reference List Pathology.pdf
-
Download Lecture Overview
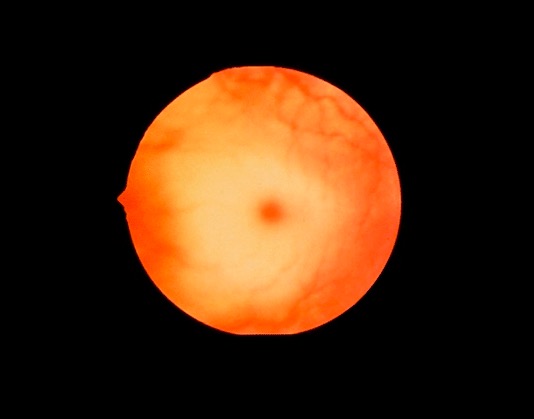
00:01 There are diseases that affect this pathway. 00:05 So, let's say we have taken up a complex structure that has multiple things in it. 00:12 Various lipids, various protein peptides. 00:15 This could be something like low density lipoprotein or this could be a food particle that has been taken up into the lysosome. 00:25 To be -- to break it down, you have to have various enzymes and various sequences to degrade each new substrate. 00:33 And eventually, you get down to elemental things, you get down to just amino acids, or you get down to just two carbon bits of triglycerides. 00:42 And that's the normal lysosome catabolism pathway. 00:45 If you have defects in any of the enzymes along the way in breaking down this complex substrate, things are gonna be -- have a bottleneck. 00:54 You're gonna be blocked and not able to go any further, and those intermediates are going to accumulate. 01:00 And that's essentially all lysosome storage diseases are. 01:06 We've lost or have defective enzymes in a sequence of degradation of some larger complex substrate. 01:14 So, an example of that, basic concept, real diseases occur because of that, Tay-Sachs disease. 01:22 So, this is an example of a lysosomal storage disease. 01:25 It's an autosomal recessive, it's lethal. It will kill you. 01:29 The clinical manifestation is that there is progressive, very early motor and cognitive deterioration. 01:36 The central nervous system is impressively affected because this disease affects the turnover of plasma -- a plasma membrane, glycolipids, that are very important for the integrity of neurons. 01:49 So, it's really gonna have most of its effect on the central nervous system. 01:54 The eyes also are very rapidly affected, so the patients will have blindness. 01:59 So, those are the kind of major clinical manifestations. 02:02 The defect is a subunit of hexosaminidase A that is responsible for catabolizing ganglioside GM2, which is a complex substrate, into one of the other additional downstream molecules GM3 or ganglioside GM3. 02:22 So, the lysosome was not able to do that. 02:24 And you can see all these other components that can be taken up, or cells are turning over their normal membranes, would need to be catabolized. 02:34 So, you can imagine that in very short order, after I show you what happens with Tay-Sachs, we're gonna see little circles with red crosses across them in all those other enzyme pathways in different diseases labeled that way. 02:47 Okay, so Tay-Sachs is specifically upstream in this ganglioside. 02:52 So, ganglioside is just a glycolipid, and it's fairly complex. 02:56 It requires multiple different enzymes to degrade it, one of the earliest ones is hexosaminidase A. 03:02 We have that defect. So, we're now no longer able to degrade it. 03:06 And so, the macrophages in the neurons accumulate that ganglioside, and then we don't recycle membrane appropriately. 03:13 And when that happens in a neuron, the neuron checks out. 03:17 And that's why you get the very rapid progressive motor and cognitive deterioration. 03:22 Okay, as promised, now, imagine all those other arrows going to other substrates. 03:28 Look at all those diseases that happen if there are enzymatic defects. 03:33 These are all lysosomal storage diseases that are real diseases that affect real people and cause real tragedy. 03:39 And you will memorize these when it comes time for the boards, but right now, it's the concept. 03:45 It's the idea that we have complex substrates that need to be progressively degraded, and there can be enzyme defects that don't allow that to happen. 03:54 And the lysosomes get big and they can potentially rupture, but you're not getting the normal turnover of the normal molecules in those cells.
About the Lecture
The lecture Resulting Condition: Tay-Sachs Disease by Richard Mitchell, MD, PhD is from the course Cellular Housekeeping Functions.
Included Quiz Questions
The catabolism of which of the following is defective in Tay-Sachs disease?
- GM2 ganglioside
- Glucocerebroside
- Ceramide trihexoside
- Sphingomyelin
- Galactocerebroside
Which of the following is associated with Tay-Sachs disease?
- Cognitive deterioration
- Peripheral neuropathy
- Cataract
- Progressive renal failure
- X-linked recessive inheritance pattern
Author of lecture Resulting Condition: Tay-Sachs Disease

Richard Mitchell, MD, PhD
Customer reviews
5,0 of 5 stars
| 5 Stars |
|
5 |
| 4 Stars |
|
0 |
| 3 Stars |
|
0 |
| 2 Stars |
|
0 |
| 1 Star |
|
0 |
