Playlist
Show Playlist
Hide Playlist
Resulting Condition: Cystic Fibrosis

-
Slides Cellular Pathology - Movement across Membranes.pdf
-
Reference List Pathology.pdf
-
Download Lecture Overview
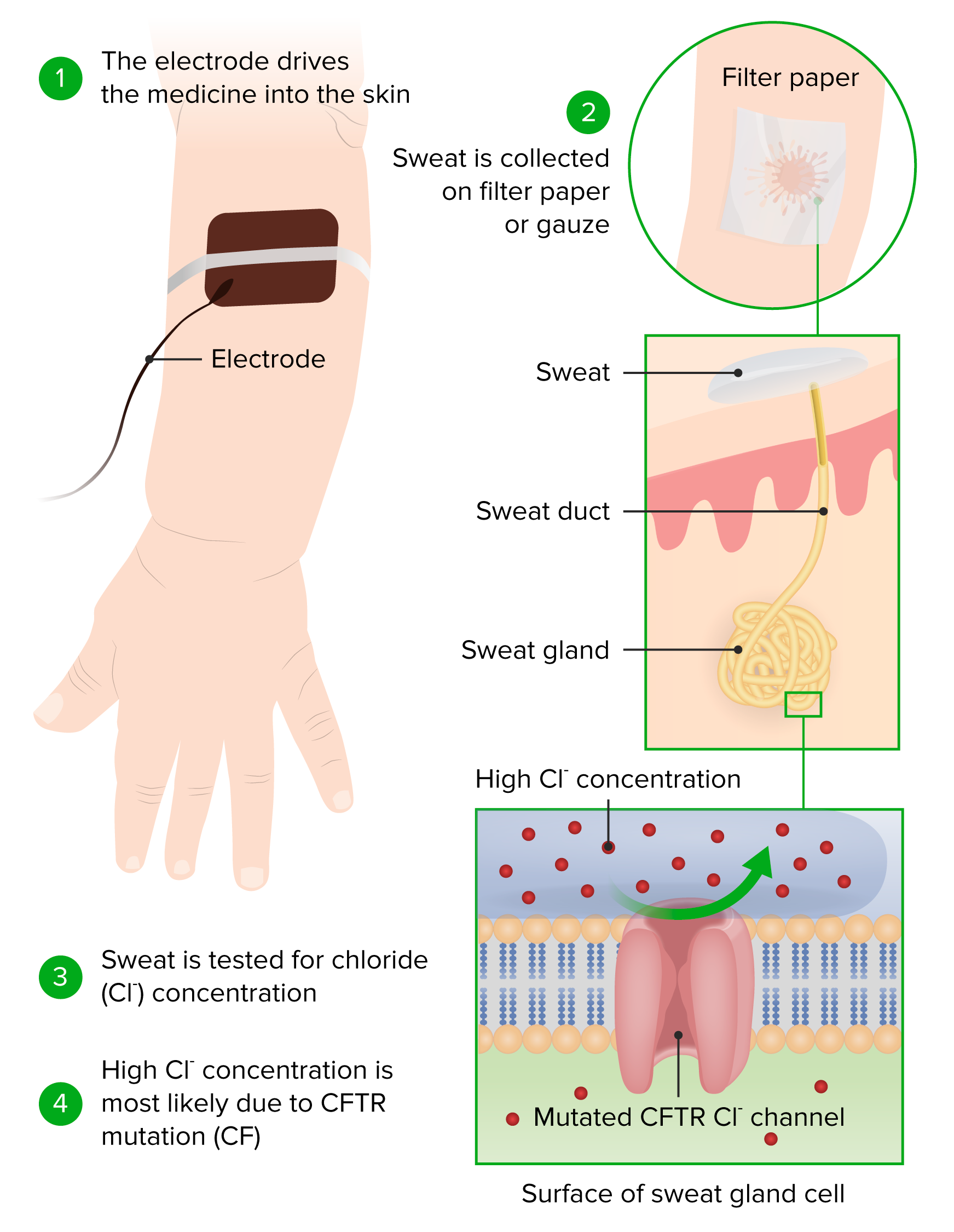
00:01 Let's look at another example where a pore protein can be mutated and cause a fairly significant disease. 00:09 And the example I'm going to use here is cystic fibrosis or CF. 00:13 CF is due to mutations in the cystic fibrosis transmembrane conductance regulator. 00:19 It's actually a very long term to describe basically a chloride channel. 00:25 That chloride channel is mutated in patients with cystic fibrosis and they do not express high levels in various epithelium. 00:35 What's happening in the right-hand picture that looks so ugly, that's actually the lung. 00:41 That is a lung that has been recurrently infected over and over and over again. 00:47 There's been massive destruction of airways, there's been massive accumulation of inflammatory cells, because of a mutation in the chloride channel that leads to thick viscous bronchial secretions. 01:02 In the next flew slides, we'll explain how that happens. 01:06 It is an autosomal recessive disorder, but in fact, may represent one of the more common autosomal recessive fatal disorders. 01:12 This lung has been taken out at transplant. 01:14 Fortunately, the patient got a new lung, but you can imagine that this is not consistent with life. 01:20 How does this work? So, the normal CFTR, meaning the normal chloride channel, is in a couple different places within the membrane of the bronchial epithelial cells. 01:32 We're looking at -- initially, the label there is the basolateral surface, and then we have the apical surface. 01:39 And there are channels on both sides that are going to be pushing ions into the cell and out of the cell. On top of the cell, we have cilia. 01:48 Those cilia are there actually to beat their part of what's called the mucociliary elevator or the mucociliary clearance mechanism that is responsible for sweeping bacteria and inhale particulate matter out of the lung. 02:03 That cilia -- those cilia sitting on top of the epithelial cells are actually sweeping a mucus layer. 02:11 And the idea is you get mucus clearance, so the arrow is pointing up towards the mouth. 02:16 That's where we would get rid of those bronchial mucus secretions that have trapped up bacteria. 02:22 We want the mucus to be a little bit sticky, because that's how you trap bacteria and other particulates that get inhaled, but we don't want it to be too sticky or too densely mucus-y, because then the poor little cilia can't sweep it out. 02:37 And this is where we get in trouble with mutated CFTR. 02:42 Let me explain a bit more about the sodium channels and the chloride channels. 02:46 So, normally, we balance how viscous the mucus layer is by pumping ions in and out. 02:53 So, sodium is, in this particular case, pumping into the membrane, across the apical membrane, and across the basolateral membrane, so that it goes to the outside world. 03:04 Potassium is also being pumped to balance the net ions. 03:10 Chloride is being pumped some low level into the space above the apical membrane. 03:18 The balance there of a sodium and chloride gives us just the right amount of water so the mucus is not too viscous. That's the goal. 03:28 We also further balance it by the movement of sodium and water that's being pumped down into the basolateral aspect. 03:36 Don't get too bugged down with all the various arrows and things like that. 03:40 Keep in mind that we want that mucus layer to be sticky, but not too viscous. 03:45 Okay, now, we have a mutation in the chloride channel. 03:50 And we are no longer putting chloride into that apical space. 03:55 And to balance that, now, we're gonna have more sodium coming into the cell and being translocated across. 04:00 And we're gonna have more sodium and water moving in the paracellular avenues to get out of the apical space into the basolateral space. 04:09 That's going to end up making a very thick and viscous mucus layer because there's less water there. 04:16 Now, this thick layer will trap bacteria, but we will not be able to sweep it out. 04:22 As a result of that, bacteria will be able to grow and can cause recurrent infections in the lungs. 04:30 And that's exactly what happens with cystic fibrosis. 04:33 So, in cystic fibrosis, we have a viscous dehydrated bronchial mucus, because just the chloride channel is not there in the amounts necessary to give us the right balance of sodium and chloride and water. 04:47 It turns out that in the pancreatic duct, we have exactly the same problem. 04:54 We don't pump chloride appropriately, and as a result, we don't have the right level of viscosity of the pancreatic secretions. 05:03 As a result of that, the pancreas auto digests. 05:07 All those pancreatic juices chew on the pancreas itself and we lose it entirely. 05:12 And those patients will develop malabsorption. 05:15 They will not be able to have any pancreas to provide the production of proteases to digest food. 05:23 We have to give them a big old pill on a daily basis to help digest their food. 05:29 The other thing that happens is it turns out that the way that the channels work in the sweat glands, if we do not pump appropriately the chloride, too much chloride and too much sodium and too much water accumulate in the sweat. 05:48 So, we end up with a very high level of sodium chloride, and it's basically salty sweat. 05:54 And moms frequently will notice that their baby, when they kiss it, is particularly salty, and that's maybe one of the earliest signs of cystic fibrosis. 06:03 So this is all about channels and carriers having disease process.
About the Lecture
The lecture Resulting Condition: Cystic Fibrosis by Richard Mitchell, MD, PhD is from the course Cellular Housekeeping Functions.
Included Quiz Questions
Which of the following is affected in cystic fibrosis?
- Chloride channel
- Na⁺/K⁺-ATPase pump
- Calcium channel
- Sodium channel
- Potassium channel
Which of the following is involved in the pathophysiology of cystic fibrosis?
- Secretion of a dehydrated viscous mucus
- Increased chloride ion secretion into the extracellular space
- Decreased mucus secretion by the mucinous glands
- Decreased sweat secretion by the eccrine glands
- Increased water secretion into the extracellular space
Which of the following is a common complication of cystic fibrosis?
- Pulmonary infections
- Renal insufficiency
- Obesity
- Cerebral aneurysms
- Peptic ulcers
Author of lecture Resulting Condition: Cystic Fibrosis

Richard Mitchell, MD, PhD
Customer reviews
5,0 of 5 stars
| 5 Stars |
|
2 |
| 4 Stars |
|
0 |
| 3 Stars |
|
0 |
| 2 Stars |
|
0 |
| 1 Star |
|
0 |
Very well explained the basic mechanism of cystic fibrosis. Hope this would lead to a cure of this disease.
The topics well explained, helped me a lot on this disease!
