Playlist
Show Playlist
Hide Playlist
Heart Failure: Pathophysiology

-
Slides Heart Failure.pdf
-
Reference List Pathology.pdf
-
Download Lecture Overview
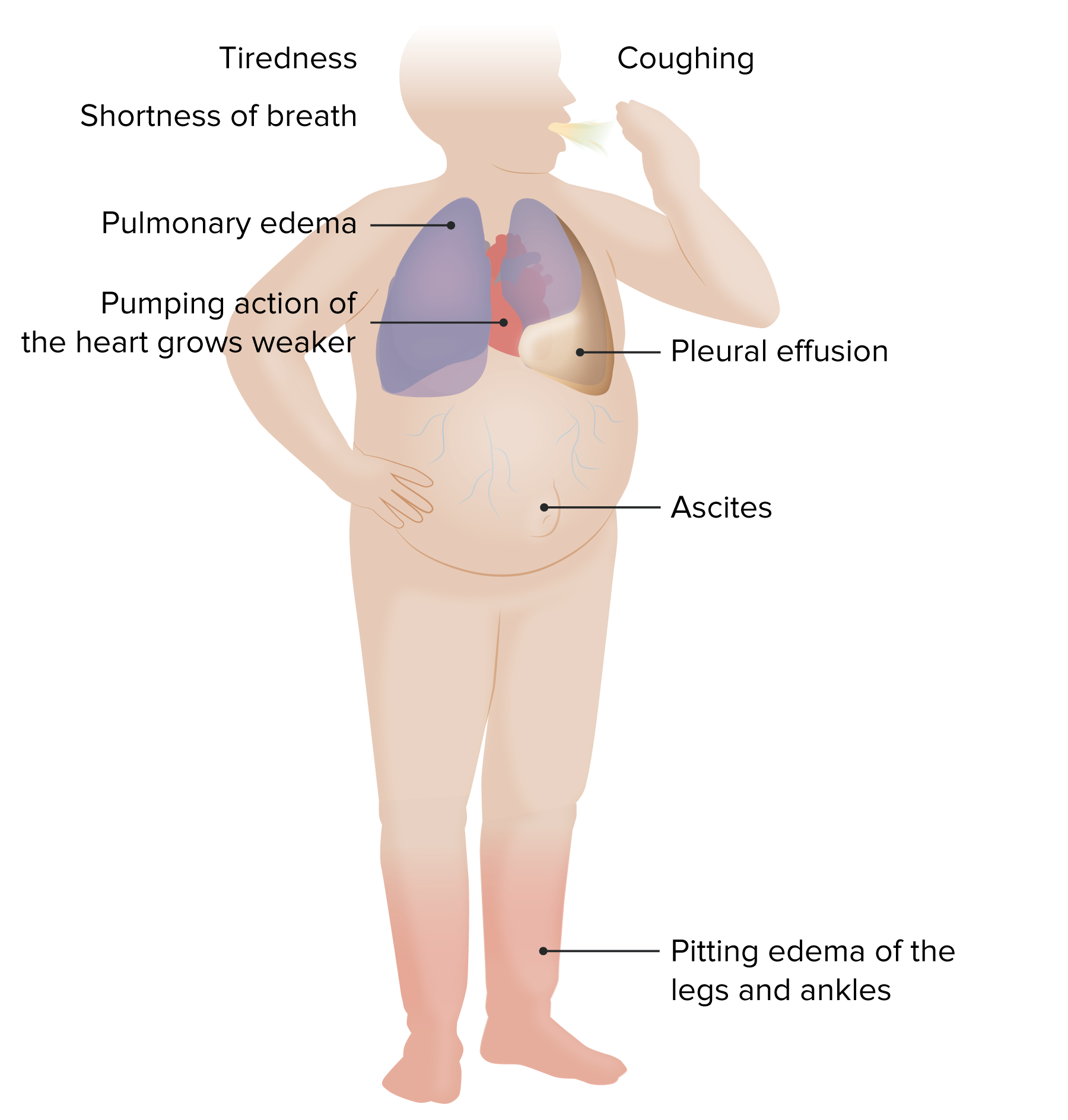
00:00 So, some of the pathophysiology associated with the heart failure. What's being indicated here is normal cardiac myocytes and we have normal flow coming in from the left atrium through the pulmonic veins into the left atrium across the mitral valve out through the left ventricle out the aortic valve and out to the aorta. If you have sustained increases in pressure, so hypertension, if you have a tight stenotic aortic valve that will not open appropriately, now we're going to increase the pressures within the left ventricle in order to provide adequate circulation. To do that, again, you can't get more cardiac myocytes, you're going to have to make them individually larger and again that may be initially adaptive but over long periods of time that may become maladaptive as we'll see on the next slide. So shown here is what it looks like histologically. The darker pinker cells down there on this H&E stain are your typical cardiac myocytes. 01:11 And as they undergo hypertrophy, they get individually larger. It's difficult to kind of draw an outline of what is a cell but I can look down at the nuclei on the normal and the nuclei on the hypertrophied myocytes and see the nuclei are considerably larger. 01:31 In order to get that adaptation to get larger myocytes, we have to increase protein synthesis making more sarcomeres, we need clearly more mitochondrion and provide greater energy, and we see this manifested as enlarged nuclei. So all those things are going up, that's great. What doesn't go up, however, are the number of capillaries. 01:55 So the capillary density in the hypertrophied myocardium don't increase in a compensatory way to provide adequate perfusion. So now I have a much larger cell with capillaries on either side and I have a relative increase of the diffusion distance into the middle of that cell. As a result of the hypertrophy, yes I get a little bit more squeeze, or maybe a lot more squeeze but now that cell can become functionally hypoxic because of the increased diffusion distance. That can lead to arrhythmias and you can also have failure of that heart muscles, it gets bigger and bigger and bigger because it's just not getting adequate nutrition. So it may undergo changes to the mitochondria, it may undergo apoptosis, you may have cell death. Not a good thing. So even in a hypertrophied heart trying to get greater output because of systemic hypertension or because of a stenotic aortic valve, you can only get so much benefit and after a while it tends to then have an adverse outcome. In thinking about the pathophysiology of heart failure, we have been talking about hypertension which is really essentially pressure overload and that can be due to either a stenotic aortic valve, for example, or could be due to systemic elevations in blood pressure. You can also have just primary valvular disease and the valvular disease depending on whether it is insufficient or stenotic will give you pressure and/or volume overload. You can also have myocardial infarction. 03:34 And basically by taking out part of the contractile force of the heart, the residual myocytes have to compensate, they have to squeeze for their lost comrades. So, any regional dysfunction, any regional infarction will potentially lead to increased demand on the myocardium. That's increased work overall through any of these various different mechanisms. With increased wall stress and the response, there are a number of adaptations that occur but the increased response is typically to have cardiac hypertrophy so we get more squeeze per contraction but we may also have chamber dilation to give us more volume. Remember where we are on the Frank-Starling curve. So we will, in general, increase cardiac size and mass. To do that, we have to make more sarcomeres, we can't make more cells. Cardiac myocytes are end-stage cells, they are terminally differentiated so we can't make more of them. But the individual cardiac myocytes can make more protein. Cool, that works. Now you have more sarcomeres. You have also the introduction of fetal gene program. So it turns out that fetal genes that occur in utero and in early life are more contractile. They actually give you more squeeze. But we turn those off as we turn into living breathing little kids and then adults and only if we need to turn on a greater contractile force we don't really have that activation of fetal gene program. That's great when it happens but those genes can also put a greater strain on the heart in an adult life and actually in the end be maladaptive. Not only are the individual cardiac myocytes feeling the effect of increased need, but the pressure and volume overload is also going to be felt by the cardiac fibroblasts and those are about 20-30% of the cells within the heart and they are going to respond not by contracting better because they can't do that but they're going to respond by making more extracellular matrix, so we're going to see more fibrosis. The problem with all these is that we've had some adaptation we haven't really changed the vasculature. So we have much larger myocytes, we've got all these other stuff going on but the capillary density hasn't changed so we can potentially have inadequate microvascular perfusion. 06:19 As a result of these changes that we talked about, you can eventually reach a point where it's maladaptive, remember the Frank-Starling curve. And you can have systolic and/or diastolic dysfunction. Systolic dysfunction when you're way out on the Frank-Starling curve you stretch everything so much, you can't squeeze better. 06:38 And diastolic dysfunction because gee we have so much fibrosis it's a stiffer heart. 06:43 So there are a variety of mechanisms at play here that will lead to the heart failure. 06:48 We've also changed the geometry, the way that cardiac myocytes interact one with another. And remember the cardiac myocytes are connected via gap junctions that allow calcium currents to go cell to cell to cell to cell. That's what allows us to have a nice contractile wave once we stimulate contraction in the first place. As we are changing all of these other features in a heart trying to adapt, we are actually changing the geometry of those gap junctions. We are also increasing potentially the ischemia because the cells are getting bigger and bigger the capillaries are not increasing in density. 07:29 And so now the individual cells are more irritable, they are more prone to dysrhythmias because we are not moving the calcium and other ions effectively because there is not enough ATP. So those are all the consequences kind of within the heart and then we're going to be flagging the entire system with the neurohumoral adaptation. 07:53 So we're going to be trying to now make the heart beat faster and beat harder and as a result of that we may actually exacerbate all these other elements of the heart failure.
About the Lecture
The lecture Heart Failure: Pathophysiology by Richard Mitchell, MD, PhD is from the course Heart Failure.
Included Quiz Questions
How does the Frank-Starling mechanism compensate for reduced cardiac output due to heart failure?
- Stroke volume changes in proportion to end-diastolic volume.
- End-diastolic volume changes in proportion to heart compliance.
- Increased heart compliance causes a decreased stroke volume.
- There is an increase in stroke volume due to myocardial remodeling.
- The neurohumoral activation of norepinephrine increases.
Which of the following leads to increased mechanical work with heart failure?
- Regional dysfunction with volume overload
- Decreased pressure
- Decreased volume
- Regional hypotension
- Myocardial stunning
What cellular change occurs in the cardiac myocytes of a patient with heart failure?
- More mitochondria
- Small nuclei
- Decreased protein synthesis
- Hyperplasia of myocytes
- No change in protein synthesis
What results from pressure and volume overload in heart failure?
- Fibrosis
- Decreased protein synthesis
- Increased heart size but a decrease in heart mass
- Angiogenesis
- KRAS mutations
Author of lecture Heart Failure: Pathophysiology

Richard Mitchell, MD, PhD
Customer reviews
5,0 of 5 stars
| 5 Stars |
|
1 |
| 4 Stars |
|
0 |
| 3 Stars |
|
0 |
| 2 Stars |
|
0 |
| 1 Star |
|
0 |
Such a great framework and categorization of heart failure pathology. Thank you Dr. Mitchell for your vision and explanation.
