Playlist
Show Playlist
Hide Playlist
Heart Failure: Introduction

-
Slides Heart Failure.pdf
-
Reference List Pathology.pdf
-
Download Lecture Overview
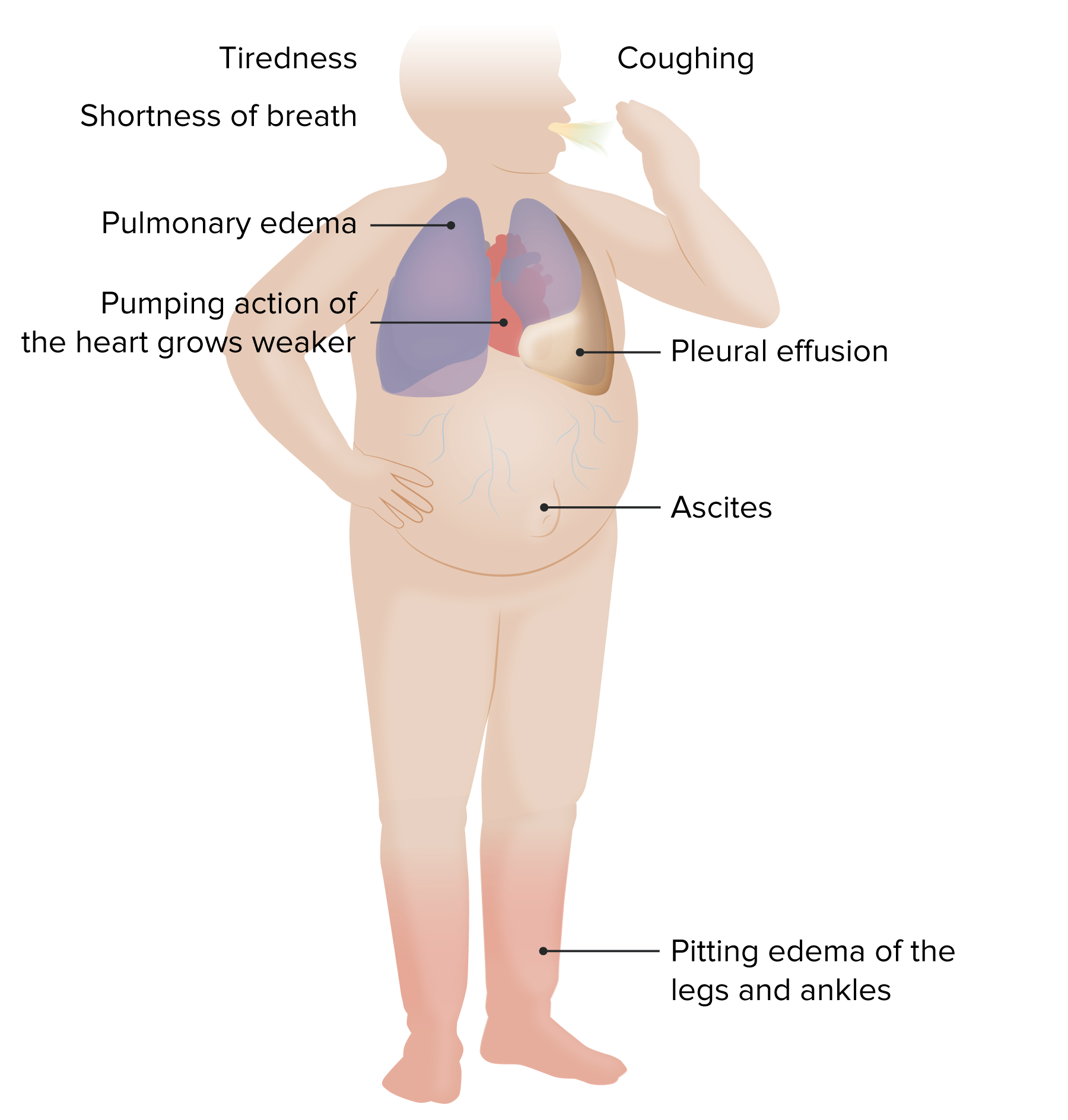
00:00 In this talk, we're going to cover a very important pathophysiologic set of consequences that lead to a lot of morbidity and mortality. This is heart failure. Fundamentally, this is pump failure and we're not able to pump blood adequately to the body and we're not able to also keep blood flowing in a unidirectional fashion so we end up with a retrograde flow which has a lot of manifestations. With that, let's get started. As I've already eluded to, the definition of heart failure is the inability of the heart to supply the body with its normal cardiac output to meet the metabolic demands. It can have a variety of causes, but that is the final important consequence. And if we're not providing adequate nutrition and oxygenation to the brain, you will have a consequence. If you're not making it to the liver, you will have problems with synthetic function. If you're not making adequate blood flow to the bowel, you're not absorbing nutrition. So there are a lot of different consequence depending on the tissue. It is the end stage of chronic heart disease not otherwise specified, meaning we can get there by a variety of different mechanisms. It is also something that can happen with acute insults. As we'll see in a couple of slides, this is a fatal disease. Heart failure that is not treated effectively will lead to death within 2-3 years and it is one of the major causes of morbidity and mortality in the world. Okay. This is showing a curve that you will see in your various physiology courses particularly in cardiovascular physiology. This is the Frank-Starling curve and on the x axis it is the ventricular filling volume that's occurring during diastole. 01:58 On the y axis is the stroke volume. That's how much the muscle squeezes during systole. As we go up to Frank-Starling curve, we are getting more and more output and we can do that by a variety of mechanisms. We can do it 1) by increasing the volume that is in the ventricle. So we can squeeze with exactly the same vigorousness and get more cardiac output. Okay, so we've dilated the heart during diastole. We fill it better. We squeeze just as well. We get more stroke volume. That's how you are able to go from resting to running a marathon. You have to increase your stroke volume. 02:37 Now we're going to come back to this Frank-Starling curve because you see it trends down and on the other side you can have maladaptation that will lead to actually diminished stroke volume as we increase the diastolic filing. Okay, so it is not something that goes up and up and up and up. We can exceed the capacity of the heart to respond appropriately. So this is just showing you one way that we can increase cardiac output, increase the stroke volume by increasing the volume that occurs during diastole. 03:11 In order to take advantage of that, we have to squeeze as vigorously as we have at lower volume filling. Okay. So, that's the Frank-Starling curve. Again there is a downside. We'll come back to that. Okay, so what is a mechanism by which we can get increased organ perfusion or increased squeeze of the heart? Well, one of the ways that we can do that is to make more sarcomeres and make for hypertrophied myocytes. 03:42 With more sarcomeres lay down in parallel next to each other, we get a concentric hypertrophy that will give us more bang for our buck. We cannot increase cell number. 03:52 Remember that myocardium, myocytes in the heart are a terminally differentiated cell and for all intents and purposes we cannot make more of them. So if we want to get more squeeze, we can increase the volume but we can increase the size of the cardiac myocytes by increasing the sarcomere content. That will give us a better squeeze. 04:17 Other ways that we can maintain cardiac function to adapt to increased demands and what is being shown here is the sympathetic trunk that lies along the aorta, the descending aorta. And there is going to be important neurohumoral mechanisms that will also help to drive cardiac adaptation if we need to increase cardiac output. Name amongst these are going to be norepinephrine release. So, if we detect diminished pressure, if we detect a decreased oxygen content, if we detect increased acidosis. 04:57 All of those are potentially going to trigger the neurohumoral system to release norepinephrine. When we do that, we'll increase heart rate and we'll squeeze more effectively. So even if we haven't made individually enlarged myocytes, this is an acute mechanism by which we can increase cardiac output. Now, we may also want to reduce cardiac output. So, whatever acute kind of stimulus has driven us to have more output, at some point we may want to reduce that again. So one of the mechanisms, there are others, but one of the mechanisms is by a volume overload of the atrium detected by the atrium as increased stretch. And that atrium makes atrial natriuretic peptide. 05:42 That peptide, a hormone released by cardiac myocytes, wow a hormone, causes increased vasodilation and diuresis. So that is a way for us to decrease blood volume and therefore reduce cardiac output. Okay, so there are these various mechanisms in play. So, the main mechanisms overall are the neurohumoral mechanisms that we've just seen and also the renin angiotensin aldosterone system, RAAS. This is going to be a system that is going to be kind of bring together the kidney, the liver, and the adrenals to maintain blood pressure. So let's start with a drop in blood pressure or a drop in fluid volume. This is detected at the level of the kidneys, at the level of the afferent arteriole in the glomerulus and you have cells of the juxtaglomerular apparatus that are going to be making a hormone renin if they detect a decrease in blood pressure. 06:52 That renin acts on angiotensinogen, a hormone secreted by the liver constitutively. 07:00 That renin acting on angiotensinogen makes angiotensin I. So it cleaves it to a smaller peptide. And then as that smaller peptide circulates around the body, endothelial cells making angiotensin converting enzyme or ACE, will act on that to make the main peptide that's responsible for regulating blood pressure. That's angiotensin II. So it's a smaller peptide. It cleaves it one more time. That angiotensin II will cause vasoconstriction. 07:32 Remember what's strict at this entire series of events was a drop in blood pressure or a perceived drop in blood pressure at the level of the kidneys. Angiotensin II will squeeze the blood vessels. So now that will raise blood pressure, but it will also act on their adrenal cortex to increase aldosterone synthesis. So here we brought together the kidney, the liver, peripheral endothelial cells, and now the adrenal and that aldosterone produced by the adrenal cortex will cause an increased reabsorption of sodium and chloride and obligate water by the kidney. Now we've increased volume. So we have tried to fix the drop in blood pressure or the perceived drop in blood fluid volume by squeezing the vessels more vigorously and by increasing the volume of the blood. Okay, those mechanisms, the neurohumoral mechanisms and the more vigorous squeezing of the heart due to hypertrophy gives us an ability to adapt to increased demands but we can go further out on the Frank-Starling curve and as we get more and more dilated, so the ventricular filling volume gets bigger and bigger and bigger we actually stretch the sarcomeres and there is a more tenuous connection between the actin and myocin chains within the sarcomeres and we do not get as much of a good contractile force. So that's the down curve on the Frank-Starling mechanism. And just because we have a bigger volume doesn't necessarily mean that we're going to have maintenance of the output. 09:16 There is a maladaptation that can occur. So there are limits to how much we can dilate up the heart in order to get more of a stroke volume. Okay, and in this particular instance we've doubled the chamber size compared to normal or non-stressed and as a result though we have stretched the sarcomeres to the point where they are no longer interacting effectively to give us the most squeeze.
About the Lecture
The lecture Heart Failure: Introduction by Richard Mitchell, MD, PhD is from the course Heart Failure.
Included Quiz Questions
What is the definition of heart failure?
- The inability of the heart to supply blood to meet the body's metabolic demands
- The inability of the heart to receive blood
- The inability of the heart to supply the lungs with blood
- The inability of the heart to coordinate a systolic and diastolic phase
- The inability of the heart to store blood
Which of the following is a mechanism to maintain organ perfusion?
- Norepinephrine release
- Systolic failure
- Neurohumoral inactivation
- Atrial natriuretic peptide inhibition
- Vasoconstriction and diuresis
What does the Frank-Starling describe?
- Relationship between stroke volume and ventricular filling volume
- Relationship between stroke volume and systole
- Relationship between diastole and ventricular filling volume
- Relationship between stroke volume and ejection fraction
- Relationship between stroke volume and vasodilation
Which of the following is an adaptation that occurs in heart failure?
- Aldosterone is released to increase the uptake of salt and water.
- Aldosterone is released to increase the excretion of salt.
- Angiotensin II causes vasodilation.
- Angiotensin II inhibits aldosterone.
- The neurohumoral system is inactivated.
What is a trigger for renin release?
- A drop in blood pressure
- An increase in blood pressure
- An increase in systolic output
- An increase in diastolic filling
- Hypocalcemia
Author of lecture Heart Failure: Introduction

Richard Mitchell, MD, PhD
Customer reviews
5,0 of 5 stars
| 5 Stars |
|
2 |
| 4 Stars |
|
0 |
| 3 Stars |
|
0 |
| 2 Stars |
|
0 |
| 1 Star |
|
0 |
A comprehensive yet simple overview of cardiac pathology provides a framework of thought and content. Dr. Mitchell has done a wonderful job of this. Thank you
It is thre first time to understan heart failur thanks for you??
